- 0
-
找原料、比价格、代采购找我!
 前小衍平台客服最近30天成交374单027-81293128
前小衍平台客服最近30天成交374单027-81293128
 产品合作与推广,清库存找我!
产品合作与推广,清库存找我! 唐令平台采购15072440602
唐令平台采购15072440602

-
您的随身业务助手
微信扫一扫 关注「前衍化学」公众号


 发询盘 看报价收询盘 拿订单
发询盘 看报价收询盘 拿订单
微信扫一扫 关注「前衍化学」公众号
发询盘 看报价收询盘 拿订单对药品杂质谱的控制是保证药品安全有效的重要措施,也是提升国产药品质量的关键环节。
自2010 年提出实施杂质谱控制的基本策略以来,经近十年持续的努力, 国内已经形成了一个比较成熟的药品杂质谱控制体系。
笔者曾对2010年之前、2010~2015 年间化学药品杂质谱控制的进展进行了综述。
2015 年以来,该领域在杂质谱控制理念、分析技术及技术应用等方面均得以迅速发展,因此本文综述2015 年以来化学药品杂质控制的进展情况,并阐述亟待解决的问题和发展前景。
对药物杂质谱(impurity profile)的控制是保证药品安全性的重要环节,也是目前国内新药研发的关键制约因素。
与杂质谱控制相关的关键技术问题可概括为:复杂体系样本的分离分析、微量组分的结构分析和微量组分的毒性评价三个方面[1]。
理想的“杂质谱控制(impurity profiling)”理念应针对药品中的每一个杂质,依据其生理活性制定相应的质控限度。
在国家重大新药创制等项目的支持下,近年来国内杂质谱控制技术得以迅速发展。
作者曾对2010—2015 年间化学药品杂质谱的研究进展进行过综述[2],本文综述了2015 年以来化学药品杂质谱研究的进展。
1法规、指导原则与应用
人用药品注册技术要求国际协调会(ICH)在制订的原料药、制剂杂质研究指导原则(ICH Q3A,ICH Q3B),残留溶剂研究指导原则(ICH Q3C)和元素杂质研究指导原则(ICH Q3D)的基础上,2014 年又颁布了基因毒性杂质研究指导原则[Assessment and Control of DNA Reactive (Mutagenic) Impurities inPharmaceuticals to limit Potential Carcinogenic Risk,ICH M7 (R1)],进一步指导创新化学药研发中的杂质研究。
虽然ICH 的指导原则对新药注册时药品中的各类杂质有了明确的要求,但如何将指导原则与具体的研发实践相结合仍有诸多问题需要探讨。
药品中的杂质可能来源于原料合成中的起始物、溶剂、催化剂、中间体、副产物等工艺过程,也可能在制剂生产、贮存和使用过程中产生。
从药品的研发至产品上市通常要经历较长的时间,不同研发阶段的关注重点应有所不同,且人们对产品中杂质的认识也是伴随着对产品工艺、生产、贮存的不断认知而深入了解。
然而,目前仅美国食品药品监督管理局(FDA)和欧洲药品管理局(EMA)针对新药不同研发阶段杂质研究的关注点进行过原则性地讨论。
Olsen 等[3]对此进行了综述。
对于工艺杂质,应在原料合成阶段重点关注产品中可能出现的各类潜在杂质;当最终合成路线确定后应重点分析杂质的去除途径,确定生产过程中的关键质控点;随着工艺过程的不断成熟,再开展未知杂质的结构确认工作,并开发新的分析方法确定是否有潜在杂质的存在。
对于手性杂质,通常需从合成工艺的角度控制各类手性异构体的产生。
在新药研发的早期,杂质的水平与产品的毒理学安全性评价结果相匹配;当新药进入临床研究阶段,产品中的杂质限度可以按ICH 的要求进行控制,也可以基于临床暴露剂量和毒理学结果适度调整;有时根据内控的安全警戒线,杂质的鉴别阈值和界定阈值可以调整至ICH Q3 的3倍,并随着临床暴露剂量的变化进行相应的调整[4];当进入III 期临床时,产品应符合ICH 的要求。
ICH M7 (R1) 对药品中的基因毒性杂质(mutagenic impurities, MIs)已经有明确的控制要求,依据毒理学关注阈值(TTC),最大的日摄入量为1.5 μg。
最初人们普遍认为在临床试验阶段也需要将MIs 控制在TTC 水平,但TTC 是基于“终身暴露时间(life time exposure)”(通常为75 年)设定的,而早期临床研究的暴露时间通常< 30 天。
业界很快就认识到这一规定缺乏科学性,并提出分阶段达到TTC 的建议[5]。
尽管ICH M7 (R1)已经允许在周期较短的临床试验中对MIs的控制可适度放宽,但这一选项并未被充分利用,而更多的是选择默认的TTC限度[6]。
这也不同程度的制约了新药研发的进程。
杂质界定(qualification)是杂质谱控制的关键环节。
由于缺少有效的界定方法,通常建议在允许的情况下应尽可能地控制杂质水平以符合ICH 的要求。
应特别关注文献中是否已有足够的数据证明已知杂质的安全性。
对特定的杂质是否需要进行界定,不仅取决于患者每日的摄入剂量,而且还与药品的适应症、给药途径、服用时间等因素有关。
虽然在新药研发中对杂质进行充分的研究是药品注册的基本要求,但对已知和潜在杂质的界定应分阶段进行。
Shaikh 等[7]从确保患者安全的角度,提出了新药研发中进行杂质界定的决策树(图1):
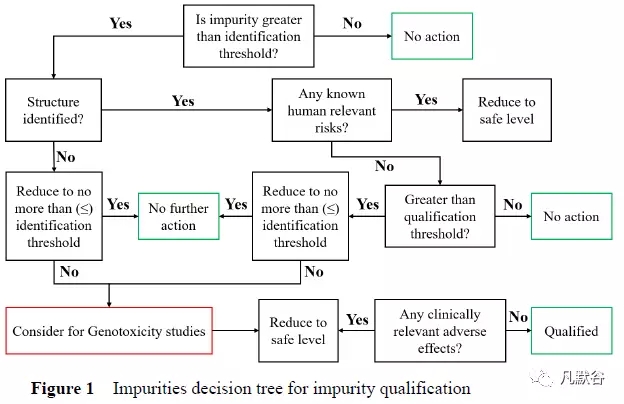
①力争将杂质水平控制在ICH 的各种阈值以下;
②对含量大于鉴别限的杂质结构进行鉴定,根据文献结果判断其可能的临床风险;
③对含量大于界定限的杂质,评估其导致临床中发生不良反应的可能;
④对含量大于鉴定限的杂质,应根据ICH M7 的要求进行基因毒性的评估。
各种烷基磺酸酯类杂质目前普遍被各国监管部门认为是磺酸盐类药物中的潜在基因毒性杂质,推测其在合成过程中与乙醇等低级醇发生酯化反应产生,因而要求企业必需对产品中是否可能残留有相应的烷基酯进行全面验证。
然而Snodin 等[8]依据烷基磺酸酯的反应机制和实验证据,认为合成工艺中形成的磺酸酯不可能达到具有显著毒理学意义水平:
从热力学角度,磺酸盐在醇中的酯化反应极难发生,需在强酸性条件下才能发生少量的转化;
在乙醇溶剂中,加入与药物碱基等摩尔的磺酸根后,二者即刻成盐,进而阻止了磺酸酯的形成;
虽然合成中更易形成氯代烷烃,但氯代烷烃极易被清除,且其在生物体内的烷基化作用较磺酸酯弱很多,根据现有的毒理学数据和ICH M7(R1)的规定,采用磺酸酯的安全限量控制氯代烷烃也是不科学的。
因此,监管部门对磺酸盐相关工艺管理方式的科学性应重新进行评估。抗体偶联药物(antibody-drug conjugates, ADCs)作为一种新兴的药物,目前ICH Q3A、Q3B和Q6B(质量标准:生物技术产品/生物制品的试验程序和验收标准)的相关规定均不能完全满足对其中小分子杂质的控制要求。
国际药物研发创新与质量联盟(The International Consortium for Innovation and Quality inPharmaceutical Development, IQ)成立专门的工作组(IWG)对该问题进行讨论[9]。
基于风险评估的方法,基于ADCs中小分子杂质的分子量、与蛋白载体的结合特性、ADC的给药浓度和给药方式等,提出了ADC中小分子杂质的安全性评估策略和构建ADC质量控制体系的方案。
ADCs中的小分子杂质不管是否已与蛋白载体结合均应进行控制;通过对ADC结合工艺的控制,减少杂质与载体蛋白的结合;通过对后续纯化工艺的控制,保证对游离杂质及药物的有效去除,使之满足ICH Q3A的一般要求;通过ADC药物稳定性的评估,预测制剂中游离小分子杂质是否能满足ICH Q3B的要求。
评价结果提示,ADC中的小分子杂质含量通常非常低,基本不会导致临床安全风险。
对仿制药技术标准的协调是药物研发的另一热点。
2018 年10 月18 日美国FDA 向ICH 提议协调全球仿制药审评标准[10]:以提高全球仿制药质量的一致性;提高监管监督效率并降低监管成本;扩大全球仿制药市场规模,通过竞争降低仿制药研发的成本,最终使患者受益。
2019 年2 月6 日,ICH 发布了对此问题的思考[11]:认为虽然许多ICH 指南适用于仿制药,但建立协调一致的仿制药注册标准具有重要意义,并将在2019 年组建仿制药讨论组(Informal Generic DrugDiscussion Group, IGDG)对其可行性进行评估。
2015 年8 月18 日,国务院印发的《关于改革药品医疗器械审评审批制度的意见》,将“提高仿制药质量,加快仿制药质量一致性评价”作为我国改革药品审评审批制度的五大目标之一;以2016年3 月5 日国务院办公厅发布的《关于开展仿制药质量和疗效一致性评价的意见》为节点,国内以生物等效为目标的口服制剂一致性评价工作蓬勃开展;2017 年12 月,CDE 一致性评价办公室又发布了《已上市化学仿制药(注射剂)一致性评价技术要求(征求意见稿)》,揭开了注射剂仿制药一致性评价的大幕。
杂质研究一直是仿制药一致性评价中审评的重点。
鉴于仿制药与参比制剂生产工艺的差异,二者的杂质谱可能不完全相同。
虽然按新仿制药的一般要求,对参比制剂中不存在的“新杂质”原则上要按照ICH 的要求进行控制,对各国药典已经收载的“已知杂质”原则上要进行鉴别与比较。
但进行一致性评价的国产制剂通常在临床中已应用了较长时间,历年的药物不良反应信息可以在一定程度上揭示仿制药的安全信息。
如何基于风险控制理念,形成我国仿制药一致性评价中的杂质评估/控制策略,是业界和监管部门面临的新挑战。
2杂质谱分析技术进展
伴随着对原料药和制剂中各种工艺杂质(包括基因毒性杂质)和降解产物监管要求的不断提高,对痕量水平杂质的表征和分析在药物杂质谱分析中越来越受到重视。
各类分析仪器的发展,特别是GC-MS、LC-MS、CE-MS、SFC-MS、LC-NMR、CE-NMR、LC-FTMS 等联用技术的发展,可实现在线对含量在~0.1%水平的杂质进行快速分析。
近年来,对已知杂质的快速识别策略已经相当完善[3,12];利用在线或离线的HPLC-MS 和/或HPLC-NMR 技术,或样品不经分离直接进行NMR 分析并结合光谱分析,对未知杂质和降解物快速进行结构确认也取得较大进展;基因毒性杂质(MIs)和手性药物对映体的分析检查也受到高度重视[12]。
2.1 MIs 分析
Teasdale 等[6]对ICH M7 实施以来MIs 的分析进展进行了系统的综述:新的分析方法更注重对一类而不是单一的MIs 进行分析,方法开发除要求具有更高的灵敏度和专属性外,还应尽量减少基质效应的干扰。
气相色谱法(GC)是分析具挥发性MIs 的首选方法,高效液相色谱法(HPLC)用于对非挥发性MIs 的分析;可以通过衍生化等方法改善MIs 的挥发性和稳定性;采用顶空进样方式可以有效避免基质效应,使得更易方法开发。
已有综述详细论述了系统分析MIs 的一般策略与方法[13, 14]。
Sun 等[15]基于MIs 的挥发性,从避免基质干扰的角度提出了选择MIs 分析方法的决策树(图2),并用于探讨稳定性实验中易形成基因毒性杂质的常见降解途径,用以指导药物的研发与审批[16]。
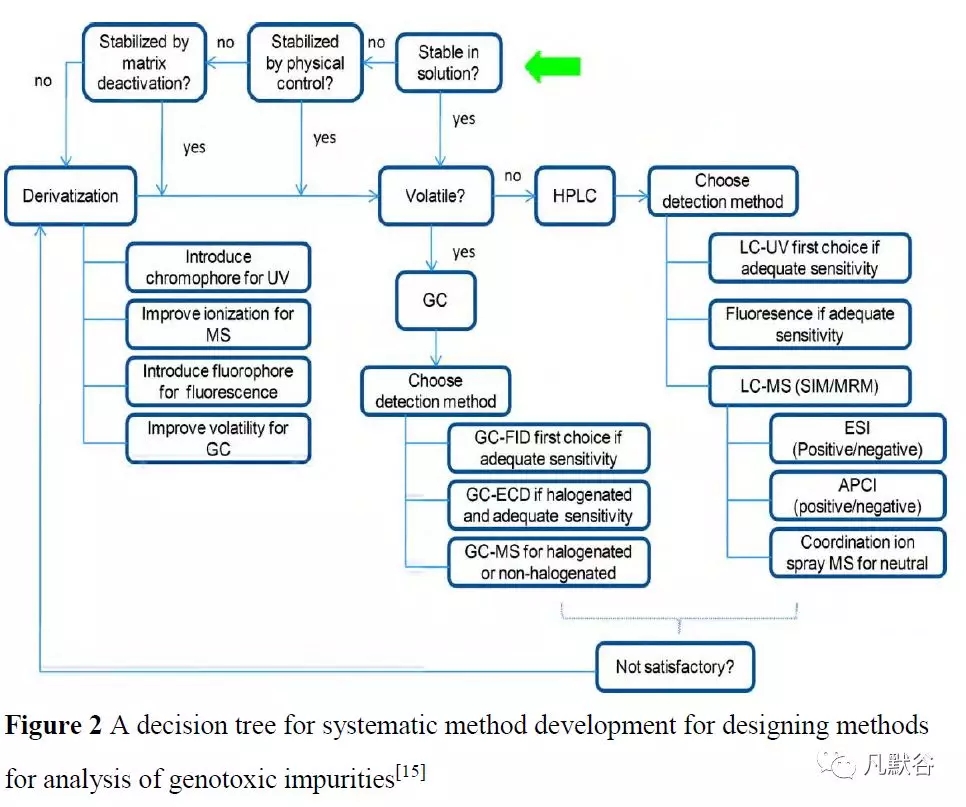
HILIC(亲水相互作用色谱法)作为其他色谱技术特别是GC 的互补方法,常用于替代GC 对极性MIs 进行分析。
McCalley[18]对HILIC 的分离机制进行了综述,可指导方法的开发。
采用HILIC-UV 法测定达伐吡啶(dalfampridine)中5 种潜在的芳香胺类MIs,其中,色谱柱的选择是关键:Zorbax 硅胶柱(5 μm)能给出理想的分析结果,其他HILIC 柱如两性柱(ZIC-HILIC 柱)或腈基柱(nitrile-HILIC 柱)的峰形较差,离子对试剂可导致方法的回收率变差;而C8或C18 色谱系统的选择性较差[18]。
利用ZIC-pHILIC 亲水作用色谱柱,采用CAD或NQAD 检测器,在含TFA 流动相中可对12 种不具紫外吸收的碱性MIs 包括已知致癌物肼(hydrazine)进行测定[19]。
Denton 等[20]利用HILIC 实现了对微量氯丙二醛(2-chloromalonaldehyde)的分析。
Douša 等[21]利用HILIC-MS 方法分析沃替西汀( vortioxetine ) 中微量的二(2- 氯乙基) 胺[2-chloro-N-(2-chloroethyl)ethanamine]。
衍生化技术可以进一步提高MIs 分析的灵敏度。
Grinberg 等在乙腈中以吡啶为衍生化试剂,对美托洛尔起始原料中的硫酸二甲酯(dimethyl sulfate, DMS)进行衍生化处理,分析衍生化产物N-甲基吡啶;
采用HILIC-ESI-MS 的SIM 检测模式,DMS 的线性范围为0.05~10 ppm,LOD 和LOQ分别为0.4 和1 ppm[22];
利用新型的衍生化试剂BPPC [butyl-1-(pyridine-4-yl)piperidine-4-carboxylate],采用HILIC-MS/MS 方法,可作为API 中烷基卤化物和烷基磺酸盐的常规分析方法或筛查方法,前者的检测水平为0.1 ppm,后者为1ppm,且方法不易受基质的干扰[23]。
离子色谱法虽然常作为 GC-MS 或HPLC-MS 的辅助技术,用于分析强极性的MIs 如烷基氯化物(alkyl chlorides)、肼等,但近年来的进展较小[6]。
Frenzel等[24]对离子色谱分析中常用的膜净化方法进行了综述;采用在线固相萃取技术,测定甲磺酸雷沙吉兰中的痕量羟胺:样品溶液中的甲磺酸雷沙吉兰被截留在IonPac CG12A 固相萃取柱上,羟胺进入色谱测定单元(CG12A 保护柱、CS12A分析柱、安培检测器);方法的LOD 和LOQ 分别为0.02 和0.04 μg·mL-1 [25]。
超临界色谱(SFC)也常作为HPLC 的互补方法。
Lesellier 和Westd 对近年来SFC的技术进展进行了综述[26]。
以超临界二氧化碳为流动相,甲醇为极性改性剂,采用两种不同的苯基柱(Synergi polar RP 和Cosmosil 5PBB),比较SFC 对多环芳烃(PAHs)分离的选择性:发现PAHs 在SFC 和HPLC 中的保留行为不同;改性剂甲醇的浓度对MIs 在Synergi polar RP 柱的分离影响较大,但对Cosmosil 柱的分离影响较小[27]。
比较SFC-ELSD 和HPLC-ELSD 分析PVC 塑料中的塑化剂(ATBC、DEHA、DEHT、TOTM),SFC-ELSD 的灵敏度更高,但HPLC-ELSD的精密性更好[28]。
毛细管电泳(CE)及电色谱技术对极性样品具有良好的分离选择性,虽然其灵敏度较低,通常不宜用于MIs 分析;但CE 的载样量高,间接紫外检测的检测限可达2 ~ 3 ppm;作为HPLC 的互补分离技术,在分析原料药中磺酸基、肼/烷基胺、叠氮化物、硫酸二甲酯和氯乙酰等PMIs 中均有应用[6]。
硼酸及相关酯类作为新的MIs,主要在一些高效偶联反应如铃木-宫浦(Suzuki-Miyaura) 反应中产生。
由于硼酸类MIs 可以通过对硼元素的测定,再通过化学计量关系得到其含量,因此,利用ICP-MS 和硼靶对样品中的残留硼进行测定,不仅灵敏度高(LOQ 为0.8 ppm,限度值为40 ppm),选择性好,且可避免常见的基质干扰。
Patel 等[29]对该方法的参数设置及优化中常见的问题进行了综述。
对烷化剂类MIs 的测定常采用GC-MS 或HPLC-MS(经衍生化处理)方法,但鉴于ICP 对硫和卤素元素的测定已达到ppb 级,这为烷化剂的分析提供了新的解决方案[30]。
利用HPLC-ICP-MS 分析烷基化剂4-氯-1-丁醇,采用3-碘苯甲酰试剂进行衍生化,方法的LOD和LOQ分别为0.2 和0.5 ppm,线性范围(μg·g-1API)为0.5~50 ppm,1~50 ppm 的准确性为95.1% ~ −114.7%,重复性(RSD)为6.2%[31]。
采用相同的方法测定烷化剂苯肼可得到相似的结果,如采用三碘代衍生物试剂,可提高方法的灵敏度,LOD 和LOQ 分别为0.06 和0.2 ppm[32]。
2.2 元素杂质分析
各类元素杂质可能在药品生产的诸环节中被无意引入终产品对患者造成危害。
ICH Q3D 建议对药品中的元素杂质应进行定性和定量限制,药品中各类元素杂质的可接受的每日接触量(permissible daily exposure, PDE)限度与给药途径有关,USP 在新颁布的通则中对药品中的24 个元素杂质提出了明确的质控要求(表1),目前国内药物研发的相关法规政策也正逐渐向ICH 靠拢。
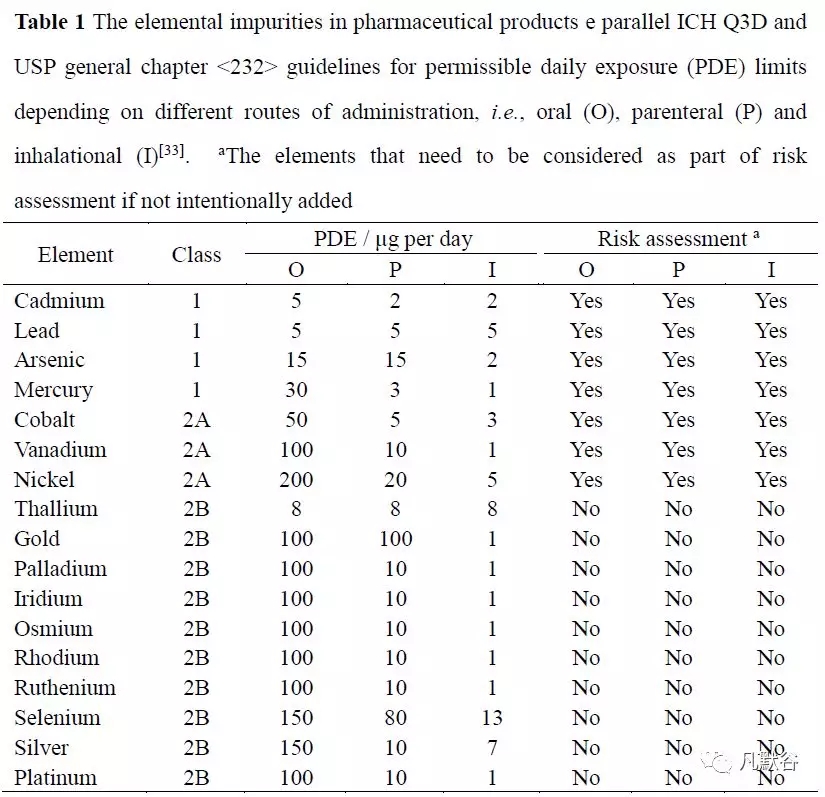
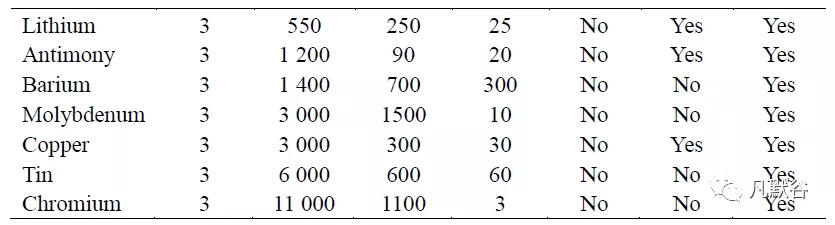
通常药品终产品中残存的痕量或超痕量的 Ir、Os、Pd、Pt、Rh 和Ru 等元素杂质,可能与原料合成中使用的催化剂有关;Cd、Hg、Ni、Pb 等元素杂质可能通过生产中的水和溶剂、合成试剂、辅料(稳定剂、填充剂、粘合剂、颜料、香料和涂料)等途径污染药物;而Cr、Cu、Mo、Ni、V 等元素杂质可能与制造过程中产品与混合罐、过滤器、填充线、包装容器等表面的接触污染产品[33]。
enke等[34]对由药品生产和包装环节引入元素杂质的风险进行了综述。
与药物接触的各类生产线、包装容器等材料,与药品接触时通常只有微量的元素实体可转移至药品中。
因此,虽然某些元素杂质在自然环境中普遍存在,但经制药过程和包装材料引入药品的风险并不高。
Boetzel 等[35]介绍了一个由制药公司联盟整理的包括201 种辅料、26 723 个检测数据的元素杂质数据库,是目前同类数据库的佼佼者,且仍在迅速扩大,可用于药品的风险评估。
Paskiet 等[36]对注射剂从常用的密封橡胶实体引入元素杂质的风险进行了评估。
电感耦合等离子体-质谱法(ICP-MS)是目前测定元素杂质的最常用手段,其次是电感耦合等离子体原子发射光谱法(ICP-AES),它们均可在多类样品基质中同时检测多种元素杂质;当仅对一种或几种特定元素杂质如Hg,、As 和Cr 进行测定时,传统的原子吸收光谱法(AAS)也可得到理想的分析结果[33]。
Wollein 等[37]采用ICP-MS、电感耦合等离子体-发射光谱法(ICP-OES)和原子吸收光谱法(GFAAS, CVAAS, HGAAS),对市场中113 个样品的21 种金属杂质进行了分析,Li 等[38]综述了31 种190 个药用辅料样品元素杂质的检测方法及测定结果,ICP-MS 特别适用于对日摄入量高的注射剂和吸入制剂中微量元素杂质的测定,定量精密度(RSD)小于4.5%;
ICP-OES 的操作较简单且具有较快的分析速度,通常可用于原材料和口服制剂中的元素杂质分析;两种分析技术的测定结果均可满足USP 通则的要求[33, 37]。
Menoutis 等[39]采用超声雾化(UN)轴向电感耦合等离子原子发射光谱法(ICP-AES)测定微量的一类和二类元素杂质,较常规ICP-MS 分析具有更低的检出限。
Balaram[40]对各类仪器分析方法包括便携式仪器、原子吸收光谱法(AAS)、X-射线荧光光谱分析(XRF)法、仪器中子活化分析(INAA)法、ICP-AES 和ICP-MS 的应用进行了综述。
采用微波辅助消解(MWAD)技术对样品进行前处理是元素杂质分析的关键环节。
USP 通则< 233 >给出了两种通用方法,分别用于ICP-AES 分析和ICP-MS分析。
Jin[41]对元素杂质分析样本前处理中的常见污染途径进行了综述,对实验室环境、试剂和器皿的控制是保证测定结果准确的关键。
Muller 等[42]通过比较浓硝酸、王水(aqua regia)和逆王水(inverse aqua regia)对4 种原料药的微波消解效率,发现硝酸和逆王水较理想,前者可有效消解500 mg 样品,后者适用于250 mg 以下样品的消解,所有的测定元素(除Os 元素易形成OsO4 影响回收外)均有较好的回收率(91% ~ 109%);
da Silva 等[43]利用逆王水建立了适用于采用ICP-OES 和ICP-MS 快速分析As、Cd、Hg、Pb 的简单微波辅助消解方法;Paskiet等[36]比较了多种对合成橡胶中元素杂质的提取方法。
2.3 浸出物/萃取物分析
药品中的浸出物一般认为是在常规或加速条件下,从与药物接触的实体(包装材料、注射器、输液管等)中迁移至药品中的化学物质;对药品中各类浸出物的分析属于杂质谱分析的一部分。
而萃取物一般认为是在实验室受控萃取研究中从试验品释放到萃取介质中的化学物质。
萃取物包括各类挥发性、半挥发性、非挥发性的有机和无机化合物,开展受控萃取研究是希望了解药品真实的浸出物谱,进而评估各类浸出物的安全风险[44]。
理想的情况是按照质量源于设计(QbD)的理念,依据药品的组成、包装系统的组成与型状、与药物的接触情况等,评估得到在生产及贮存过程药品中浸出物的安全空间;Jenke以塑料包装的注射液为例,论证了该理念的可行性[45]。
从药用聚合物材料萃取物中鉴定出的540 余种化合物的毒理学信息包括未观察到作用的水平(no observed effect levels, NOELs)、未观察到有害作用的水平(no observed adverse effect levels, NOAELs) 、公布的最低中毒剂量(lowestpublished toxic dose, TDLOs)等毒理学终点指标已被汇总,可用于浸出物的风险评估[46];
由一个非营利性联合体-产品质量研究所(PQRI)设立的浸出物/萃取物工作小组,对药品研发过程中与浸出物/萃取物有关的科学和管理问题进行了探讨,目前已在浸出物/萃取物分析的标准化工作程序和安全阈值等方面达成共识,且分别用于对口腔吸入剂、鼻腔制剂、注射剂、眼用制剂中的浸出物的风险评估[47,48]。
采用水和有机萃取溶剂对生产注射剂包装袋的21 种常用聚丙烯树脂进行萃取研究,并根据萃取物谱评估其对注射剂长期、大剂量治疗时的潜在风险[49]。
虽然由于处方工艺的差异,在不同萃取条件下不同的聚丙烯树脂的有机萃取物谱差异较大,但依据特定的萃取成分如抗氧剂等可将其分为不同的组,同组样品的萃取物谱具有相似性;大多数萃取液中元素杂质(铝、硅、碱金属和碱土金属)的量均较低。
浸出物/萃取物分析的另一关键点是如何保证所有浸出/萃取出的物质都得到检出。
色谱法是常用的分析方法。
Jenke 等[50]采用互补的色谱系统如GC 和HPLC结合多种检测方式,分析药物包装、生产和释药系统等常用的塑料材料中的萃取物谱,为防止色谱分析中遗漏了某些萃取物,利用总有机碳含量(TOC)测定法评估对各类水提物(包括缓冲液提取物)分析结果的完整性,并在对无菌滤器水提物等的分析中得到了较好的应用。
对提取物中不适合用GC 进行分析的非挥发性及热不稳定化合物,在由大气化学电离质谱结合紫外检测器组成的UPLC 系统中,22 min 即可实现对多种常见提取物中的代表性化合物混合物进行充分的分离与检测,在对实际萃取样品进行分析时,即使分析对象是不相容的有机萃取液,也不会对色谱系统产生显著影响[51]。
通过模拟浸出(迁移)试验(由低密度聚乙烯瓶、聚丙烯瓶盖和胶塞等组成、萃取溶剂分别为pH 2.5 的缓冲溶液、pH9.5 的缓冲溶液和体积比1:1 的异丙醇/水),揭示注射剂和滴眼剂包材的浸出物谱。
包材的萃取物谱与包材的化学组成与结构密切相关,且受萃取介质和具体浸出物化学性质的影响;虽然药物与包材直接接触可能加速某些浸出物的迁移,但并不是发生迁移和浸出的先决条件[52]。
胶塞中的析出物与头孢菌素相互作用,导致贮存过程中药品溶解时逐渐变浑浊是国内突出的质量问题[53]。
利用GC-MS 建立的胶塞挥发性成分分析数据库,不仅可以快速分析常用胶塞中的主要挥发性迁移物,还可以快速确认易与头孢菌素相互作用的迁移物;结合模拟吸附试验,可形成有效的头孢菌素-胶塞相容性试验策略,进而针对性地建立胶塞质控方法[54]。
2.4 AQbD 理念及应用
风险管理理念与质量保证体系的紧密结合是保证药品质量的重要环节。
杂质谱控制过程中,保证杂质质控分析方法在整个药品生命周期都具有良好的专属性和敏感性。
分析方法质量源于设计(analytical quality bydesign, AQbD)理念作为一种面向风险管理的方法论,近年来在建立杂质谱分析方法时被广泛接受。
与传统的质量源于检测(quality by testing, QbT)方法相比较,其根据分析目标的变化范围(analytical target profile, ATP),利用实验设计(DoE)的方法,同时考虑管理与分析方法的风险来确定设计空间(design space,DS),可最大程度的保证方法的有效性[55, 56]。
采用AQbD 理念建立的分析方法,由于允许实验参数在操作设计区域(method operable design region, MODR)内变化,方法具有更好的粗放性,可减少实验中的超出趋势结果(out of trend, OOT)和超出标准结果(out of specification, OOS)[57];
同时,由于可最大限度地减少在方法转移(transfer)、性能确认(verification)和方法变更中的工作量,有助于降低对分析方法生命周期管理包括方法设计、方法开发和方法验证(仪器的检定、持续的方法性能验证和方法转移)的成本[58]。
Dispas 等[56]对近年来AQbD 在杂质谱分析中的应用进展进行了综述。
在基于AQbD 理念的HPLC 方法开发中,化学计量学方法在确定适宜的设计方案筛选关键影响因子,建立定量关系模型确定方法的操作空间等方面,发挥着越来越重要的作用(图3)[59]。
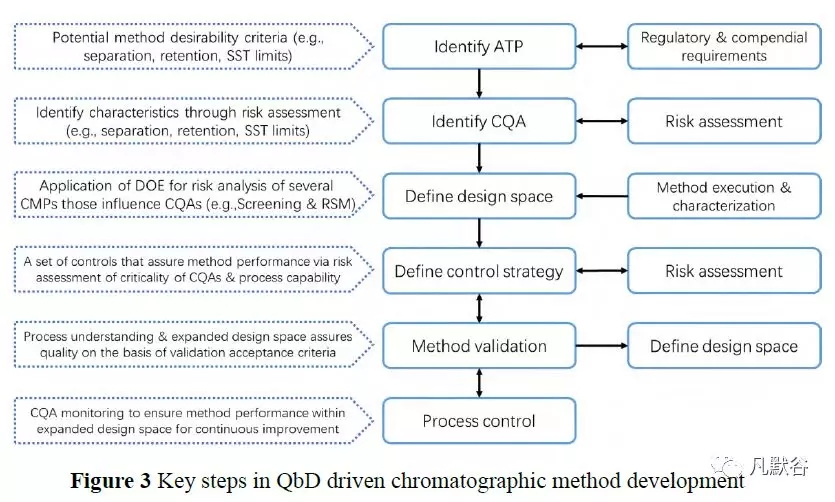
Tumpa 等[60]提出了基于QbD 的理念建立HILIC 方法的指导原则;Zhang 等[61]利用混合过程变量(mixture-process variable, MPV)设计,建立了基于QbD 理念的HPLC 双梯度洗脱分析方法,用于氯唑西林稳定性试验中降解杂质的测定;Taheri 等[62]在对塞来昔布(celecoxib)共洗脱物的半制备分离中,利用中心复合设计成功的选择出最佳色谱分离条件。
对利用 HPLC-MS/MS 等联用技术建立的可同时测定几十至几百种目标分析物的分析方法,如对残留农药等的测定,按传统的方法验证要求,通常需要采用标准加入法对定量准确性等参数进行验证。
该验证程序不仅费时、费力;且当目标分析物变化如增加了新的控制对象时,需重新对方法进行验证;实验中如对照品的加入量不适宜,还可能得到不正确的验证结果。
Alladio 等[63]利用化学计量学方法,建立了对新目标分析物的保留时间、基质效应、回收率、LOD 和LOQ进行预测的偏最小二乘法模型,用于评价已有方法对新目标分析物的分析能力,并取得了预期的评价效果。
利用定量结构-保留关系(QSRR)不仅可以预测分析物的保留时间,且有助于对其分离机制的了解。
对未知组分保留值的预测可以帮助选择分析方法,减少方法开发的时间。
Amos 等[64]对建立QSRR 模型的方法、关键点和预测精度等进行了综述。
根据溶质的色谱相似度指数(chromatographicsimilarity index)建立局部QSRR 模型可准确预测溶质的保留值[65]。
比较4 种常用的评价QSRR 模型预测准确性的表征方法,认为预测均方根误差百分比(RMSEP)是对QSRR 模型预测能力的最佳估计值[66]。
利用定量结构-保留关系(QSRR)模型,通过预测分析物在5 种不同HILIC 固定相上的保留值,可以帮助选择最适宜分离的固定相[67]。
利用定量结构-保留关系(QSRR)结合疏水消除模型(HSM),预测分析物在反相液相色谱系统(RPLC)中的保留值,可以预测药物杂质是否与药物活性成分(API)的共洗脱[68]。
当缺乏杂质对照品,无法判断一个新建立的色谱方法是否可以对药物中所有的已知杂质都能检出时,利用QSRR 模型,通过预测已知杂质的保留时间,可以帮助判断方法对已知杂质的检出能力[69],并可以预测新杂质的色谱行为[70]。
2.5 其他
选择适宜的色谱柱依然是药物杂质谱分析的热点。
不同色谱柱的选择性差异,常导致无法重现文献甚至药典中收载的成熟的杂质分析方法。
因此,如何快速寻找到性能适宜的色谱柱常成为实验的关键。基于疏水消除模型的色谱柱表征体系已在色谱柱的选择包括选择相似或互补的色谱柱,选择最佳分离色谱柱中广泛应用[71],并推动了色谱柱选择模式的发展[72-74]。
基于疏水消除模型,发现影响β-内酰胺类抗生素难分离杂质对分离的关键色谱柱参数是柱参数A,并提出了参数A 的最佳区间[75]。
以克拉霉素杂质分析为例,选择适用于分离难分离杂质对的色谱柱,提出了应用疏水消除模型针对特定分析方选择最佳色谱柱的策略[76]。
上述研究体现了色谱柱选择理论在实用化方面取得的进展,而通过分子模拟方法如分子动力学或蒙特卡罗方法,最大化地减少实验筛选过程是色谱柱选择应用研究的目标[77]。
药物杂质谱分析中的另一关键点是对杂质的检测与定量。在对各类不具有UV吸收的样品的分析中,基于气溶胶(aerosol-based)检测的各类通用性检测器特别是电喷雾检测器(charged aerosol detectors,CAD)和电化学检测器发挥着越来越重要的作用。
虽然CAD检测器被普遍认为是一种质量型检测器,可以在没有对照品的情况下实现对溶质绝对含量的准确估计,然而对50种具有广泛物理化学性质的化合物的HPLC-CAD测定结果表明,相对于传统的质量校正定量方法,通过估计检测颗粒的相对表面积进行校正,可得到更准确的定量结果,特别是可以明显提高对密度较高溶质定量的准确性(与NMR定量的平均误差为5.8%)[78]。
在杂质谱分析中利用杂质对照品进行定性与定量分析是最理想的方案,在药典等质量标准中,杂质对照品的使用也越来越普遍。
与此同时,对杂质对照品的制备、供应等全生命周期的管理要求也越来越严格[79]。
我国在2016年5月施行的《化学药品新注册分类申报资料要求(试行)》中,对不同注册分类的药品包括原料和制剂研制过程中使用的对照品(包括主成分对照品和杂质对照品)需提供相关资料。
对于药典对照品,不仅需提供批号、纯度、说明书等详细信息,还要提供来源证明;研制过程中如果使用了外购对照品或自制对照品,除需提供来源证明外,还需提供结构确证、质量标准以及含量标定过程等信息。
如何协调各国监管部们对杂质对照品的要求,提高各类杂质对照品的可及性是杂质谱研究的另一关键点[80]。
采用混合杂质对照品进行定性[80,81],采用加校正因子的主成分自身对照法定量,是解决方案之一。当
杂质与主成分的校正因子在0.9~1.1,可直接采用主成分自身对照法计算含量;当杂质与主成分的校正因子相差较大时,利用校正因子消除杂质响应值差异对测定结果的影响。根据质量平衡原理,利用HPLC-DAD结合ELSD和MS分析,可以保证校正因子测定的准确性[82]。
然而,利用定量NMR结合HPLC分析是消除校正因子测定中由杂质对照品含量不准确引入的误差的最方便有效途径[83]。
3杂质分析技术应用进展
药物中的杂质包括工艺杂质和降解杂质,基于药物的结构、来源、工艺等特点,从整体上思考一类药物的杂质谱分析策略,是杂质谱控制的一个飞跃,对产品的质量控制具有更实际的意义。
王丽娟等[84]对二氢吡啶类药物的杂质来源、检测方法、杂质限度以及评价标准等相关内容进行了综述;袁耀佐等[85]回顾氨基糖苷类抗生素组分/杂质分析技术的发展历程,讨论了常见氨基糖苷类抗生素组分/杂质分析方法的优缺点,进而分析应用液相色谱脉冲安培电化学检测方法在氨基糖苷类抗生素组分分析的优势,探讨该类药物未来质控的发展方向。
3.1 杂质谱分析方法
如何明确药品中的杂质组成进而建立适宜的分析方法是杂质谱分析首先需要解决的问题。
在国内根据特定品种在药典标准中收载的具体杂质结构,结合文献报道结果,对已有杂质谱分析方法进行改进与优化,依然具有较好的实用性。
如对塞来昔布[86]、盐酸西那卡塞[87]、杆菌肽[88]原料药、克霉唑乳膏[89]和复方依折麦布瑞舒伐他汀钙片[90]有关物质HPLC分析方法的改进等。
而根据原料合成及制剂工艺的特点,建立适宜的杂质谱分析方法,对生产工艺进行评价与控制则成为新的热点。
如对安塞曲匹[91]、富马酸沃诺拉赞[92]、呋布西林钠[93]原料及盐酸伐地那非口腔崩解片[94]有关物质的分析等。
药物稳定性试验作为确定药物中降解杂质的重要环节,强制降解试验作为预测药物长期稳定性的重要工具,可以了解药品的可能降解途径和降解产物等,进而指导建立杂质谱分析方法[95]。
通过对稳定性试验方案的合理设计,不仅可以评价药物处方的合理性,确定产品的有效期、复验期和贮存条件等,满足监管的需要,且稳定性试验中产生的降解杂质还可以用于验证杂质谱分析方法的有效性[96]。
如通过对氨曲南稳定性实验中3个新未知降解杂质的鉴定,建立了新的杂质控制方法[97]。
利用强制降解试验还有助于在杂质谱分析中对杂质的结构进行确认,如利用强制降解试验分析利奈唑胺片[98]、头孢拉定二水合物[99]、紫杉醇及其注射液[100]的杂质谱等。
此外,通过对不同厂家生产的注射用头孢西丁钠杂质谱及杂质降解途径异同的分析,并结合强力破坏试验对样品中主要杂质的来源和降解途径进行研究,可以更全面地了解产品的特性,进而完善药品质量标准[101]。
在国家评价性抽验中,通过对市场中不同企业相同产品的杂质谱的系统比较,可以较好地掌握同品种药品制剂中杂质谱的组成等信息,进而建立理想的杂质谱分析方法。
如对多肽类药物醋酸奥曲肽原料及其制剂[102]、半合成抗生素依托红霉素片[103]与醋酸麦迪霉素颗粒剂[104]、化学药口服制剂普伐他汀钠及其制剂[105]、缬沙坦胶囊[106]、非甾体抗炎药酮咯酸氨丁三醇注射液[107]、顺铂注射剂[108]和利多卡因气雾剂[109]等有关物质分析方法的建立;而采用试验设计理念对复杂成分的样品如丙酸交沙霉素进行方法优化,可获得更理想的分离结果[110]。
而对不同企业产品杂质谱的比较,可用以评价产品质量的差异,揭示生产工艺与杂质谱的相关性。
如对硫酸依替米星[111]、注射用磺苄西林钠[112]、和注射用头孢西丁钠[113]等的质量评价。
将杂质谱分析与生产过程相关联,则可以更好地理解生产工艺与杂质谱的关系,进而实现精准地对关键生产工艺的控制。
如阿莫西林克拉维酸钾片剂杂质谱分析结果揭示,制剂过程中阿莫西林三水合物的脱水与阿莫西林闭环二聚体的产生呈正相关[114];头孢拉定颗粒杂质谱分析结果揭示,对制粒前温湿度的控制是工艺控制的关键[115];注射用盐酸甲氯芬酯杂质谱分析结果揭示,杂质A为水解杂质,主要由制剂冷冻干燥过程引入,杂质B 和杂质C为醇解杂质,主要由原料药合成工艺引入,与工艺中使用的低级醇类有关[116]。
而注射用氨苄西林钠氯唑西林钠杂质谱的差异主要与氨苄西林钠原料的生产工艺有关,冷冻干燥工艺的氨苄西林钠原料较溶媒结晶工艺产品更不稳定,制剂更易受温度等环境因素的影响[117]。
选择适宜的手性色谱柱常规HPLC方法,对实际产品中的手性杂质进行分析,依然是药品质量控制的常用方法。
如采用硅胶表面涂布直链淀粉-三(3,5-二甲苯基氨基甲酸酯)为填充剂的手性色谱柱,对替格瑞洛中异构体杂质[118]、枸橼酸托法替尼原料及片剂中对映异构体杂质[179]的测定等。
然而采用超临界流体色谱法对药物中对映异构体的分析是近年来的研究热点。
如对1,4-二氢吡啶类钙拮抗剂(尼索地平、西尼地平、普拉地平、阿折地平、盐酸马尼地平)的手性拆分[120],对依折麦布及其R-对映体的手性拆分[121]等。对多肽类药物的杂质控制研究也受到关注。
如以多肽合成中的中间体、潜在副产物和降解产物等为研究对象,建立醋酸亮丙瑞林(一种合成的线性九肽)有关物质分析方法[122];采用LC-MS分析鲑降钙素注射液中的降解杂质[123];采用离子色谱对糖肽类药物中游离糖含量的分析[124]等。
对氨基酸类、糖类和酯类等结构相对简单且不具有UV吸收的药物/辅料的杂质分析研究的进展较缓慢,虽然有采用超临界流体色谱质谱联用技术定性分析中/长链结构甘油三酯组成[125]的报道,但主要研究仍采用HPLC紫外末端检测,如测定门冬氨酸钾镁注射液中门冬氨酸的有关物质[126]、盐酸氨基葡萄糖的有关物质[127]等;如引入新分离机制的色谱系统如HILIC等,结合新型的检测器如CAD、NQAD等,有望促进该领域的发展。
对b - 内酰胺抗生素中聚合物的分析,目前中国药典收载的Sephadex-G10凝胶色谱系统已经无法满足现代质控的需要,而基于高效凝胶色谱技术方法的专属性也越来越受到挑战[128]。
由于二维色谱技术的发展,利用柱切换技术可以方便地将凝胶色谱系统中的色谱峰在常规HPLC色谱系统中进行定位,结合LC-MS技术即可在杂质分析方法中将聚合物杂质作为特定杂质进行控制[128,129],这也将成为控制 -内酰胺抗生素聚合物的发展方向。
3.2 微量杂质结构解析
采用LC-MS技术对产品中的工艺杂质和降解产物等微量成分的结构特征进行分析,依然是目前识别杂质结构的主要方法[130],对同系物药物及杂质裂解规律的探讨有助于对杂质的识别。
王聪等[131]对7个β-受体激动剂的电喷雾质谱裂解规律进行了探讨,Wang等[132]对b - 内酰胺抗生素和其开环物的质谱裂解差异进行了比较,Qian等[133]对头孢克洛和其△3异构体的质谱裂解差异进行了比较;而NMR技术及联用技术在杂质定性分析中的应用也越来越广泛[134]。
国内近年来在对结构复杂且存在手性中心的未知杂质进行鉴别时,普遍采用制备分离,再利用各种波谱技术对其结构进行推测的策略。
如对盐酸多西环素[135]和利福拉齐[136]杂质等的鉴别。
而利用高分辨质谱,根据一级、二级质谱裂解规律推测化学药品中杂质的结构已逐渐成为主流。
如对尼麦角林[137]、碘海醇中有关物质O-烷基化合物和N-烷基化合物的准确分子量和结构推测[138],采用电喷雾离子化正负2种离子模式鉴定达比加群酯中间体[139]和瑞舒伐他汀中间体[140]中的杂质,采用飞行时间质谱推测佐匹克隆[141]、盐酸左西替利嗪片[142]中的杂质结构;采用高分辨飞行时间质谱分析氯沙坦钾[143]和创新药物艾托莫德[144]中的未知杂质。
二维液相色谱-质谱联用技术则解决了色谱流动相不适宜直接用于质谱分析的难题。
如用于分析来氟米特片的杂质谱[145]和对咪达唑仑注射液中杂质结构的解析[146]等。
3.3 微量/痕量杂质分析
对微量毒性杂质和MIs的分析近年来的发展较快。
沈雨婷等[147]采用UPLC-MS/MS方法,测定了盐酸哌替啶原料及注射液中的神经毒性杂质1-甲基-4-苯基-1, 2, 3, 6-四氢吡啶;栾绍嵘等[148]利用离子色谱安培法检测维生素B6与2-噻吩乙酸中的氰化物;周婷等[149]利用乙醇衍生顶空气相色谱法测定氢氯噻嗪原料药中微量甲醛和甲酸的含量;采用HPLC柱前衍生化法测定聚山梨酯类辅料中醛类物质[150]。
对MIs的检测是当前国内的热点。
刘晓丹等[151]对MIs在医药工业中的可能来源等进行了分析;张霁等[152]对药物研发中MIs的控制策略与方法进行了综述;刘雪薇等[153]综述了药物中磺酸酯类MIs的研究进展;谢含仪等[154]对MIs分析方法和前处理技术的研究进展进行了综述。
虽然采用HPLC-UV方法有时可满足MIs常规质控的要求,如对利伐沙班[155]、间苯三酚[156]中苯胺类MIs的分析等,但为提高方法的灵敏度与准确性,更多的是采用HPLC-MS和HPLC-MS/MS方法,如对酒石酸伐尼克兰[157]和氟胞嘧啶[158]中痕量苯胺类MIs的测定;
对硫酸氢氯吡格雷[159]和草酸右旋西酞普兰[160]中甲苯磺酸酯类MIs的测定;
对奥拉帕尼中氨基甲酸酯类MIs的测定[161];
对阿哌沙班中的3-吗啉基-1-[4-(5-氯戊酰胺基)苯基]-5,6-二氢吡啶-2(1H)-酮(2)[162]、卡非佐米中的(2S)-2-氨基-4-甲基-1-[(2R)-2-甲基环氧乙烷基]-1-戊酮(3)[163]等MIs的测定。
然而,如何将针对具体品种的测定方法形成通用的检测/筛查平台是亟待思考的问题。
在元素杂质分析方面,徐文峰等[164]综述了电感耦合等离子质谱(ICP-MS)在药物分析中的应用。
采用微波消解-ICP-MS法,可对蚓激酶[165]、盐酸安舒法辛缓释片[166]中的多种有害元素同时进行测定。
ICP-MS法还可同时测定玻璃类药包材中的多种迁移离子,有助于开展相容性及对玻璃药包材的质控。
如注射用泮托拉唑钠在低硼硅玻璃管制注射剂瓶中,经强降解试验、加速试验和长期试验后,发现产品中的硅、钙有上升趋势,铝、硼也有一定程度的上升,镁、镉、铅、砷、锑迁移数值略有波动,但均明显低于PDE值要求[167];采用4%的醋酸溶液对玻璃药包材进行浸提,测定浸提液中的11种元素杂质,发现铝的浸出量较高,澄清剂氧化铈应用广泛,建议对铝、铈的浸出量作为玻璃药包材的检测指标[168]。
在对各类药品包装材料中浸出物/迁移物的分析方面,系统的研究尚较少见。
付蒙等[169]采用GC-MS分析卤化丁基胶塞中的挥发性成分和可提取物信息,认为挥发性成分中的寡聚物和抗氧剂可以反映生胶的种类与来源,环硅氧烷类化合物可以体现不同胶塞的硅化工艺差异,其他挥发性成分如饱和烷烃类与胶塞具体配方有关,可提取物成分中的S8,可用于部分硫化体系的识别。
其他较多的研究集中在对各类残留物、添加剂,特别是抗氧剂迁移量的检测上。
如采用GC-MS/MS测定一次性使用输液器产品中增塑剂偏苯三酸三辛酯(TOTM)的溶出量[170]、采用LC-MS/MS测定镀膜胶塞中全氟辛基磺酸(PFOS)和全氟辛酸(PFOA)的迁移[171]、采用GC测定胶塞环硅氧烷类化合物的迁移[172]及对三层共挤输液用袋[173]、塑料输液包装材料与容器[174]、药用丁基胶塞[175]中的各类抗氧剂等的测定。
而利用GC-MS法在盐酸金霉素眼膏中发现多种多环芳烃[176]和多种邻苯二甲酸酯[177],提示对凡士林辅料中的各类迁移物的控制势在必行。
3.4 杂质的毒性评估
对化学药品中毒性杂质的识别和判定一直是杂质安全性评价和限度控制的难点。根据化学药品杂质毒性的性质,目前可将其分为3类:MIs,普通毒性杂质和普通杂质。
对MIs,使用杂质纯品,利用体外细菌致突变试验(Ames试验)对其进行评估是理想的方法,但鉴于实际应用中的困难,FDA和欧洲药品管理局(EMA)等提出,在对杂质结构进行鉴定的基础上,可利用定量结构-活性关系(QSAR)模型,首先通过计算机预测杂质是否具有“警示结构”;
如果计算机预测该杂质不具有“警示结构”,则可判断该杂质不具有基因毒性;
如果判断该杂质具有“警示结构”,则可通过Ames实验进行试验确认,若Ames实验结果为阳性,该杂质按MIs控制;
若为阴性,则判断其不具有基因毒性[178]。
对非遗传毒性杂质,通常希望对其是否具有特定的毒性作用进行进一步的评估,以便在杂质谱控制中确定是否将其作为特定的毒性杂质进行控制。
利用计算机(in silico)预测杂质与API的ADMET特性的差异,对杂质的毒性作用进行评估是一种快速有效的方法,如对罗氟司特(roflumilast)[179]、盐酸头孢替胺(cefotiam hydrochloride)[180]诸降解杂质的评估。
近年来中国食品药品检定研究院与中国医学科学院医药生物技术研究所合作,完善了斑马鱼药物杂质毒性评价平台:利用LC-MS/MS定量测定头孢菌素在斑马鱼体内的吸收情况[181],并进一步通过QSAR方法,建立了斑马鱼体内吸收与药物结构之间的定量关系模型,预测其他头孢菌素及杂质在斑马鱼体内的吸收情况[182];利用胚胎毒性试验中的致畸率、致死率表征杂质的急性毒性,利用毒性表型的差异说明杂质毒性[183];
利用幼鱼运动行为(游动距离、游动速度、对光刺激的反应)的改变,表征杂质对机体神经系统的作用[183,184];
以幼鱼心跳速率的改变,结合对心脏发育畸形的观测[183,185],或成鱼心电图的改变表征杂质对心脏功能的影响;在此基础上总结出头孢菌素结构与急性毒性、神经毒性、心脏毒性等的关系,成功地对国家创新药物头孢嗪咪钠中杂质的毒性进行了预测[186],形成了系统地对头孢菌素杂质毒性评价的策略与方法[187]。
此外,完成了对喹诺酮类药物结构与神经毒性等关系的探讨[188]。
在此基础上,又将系统毒理学研究的策略和方法应用到对杂质的毒性评价中:通过生物信息学方法,筛选药物与杂质的差异表达基因(DEGs)和差异共表达基因(CDEGs);
构建药物-基因、药物-信号通路分子调控网络,探讨特定的靶器官毒性及作用机制;构建蛋白-蛋白相互作用网络,寻找药物毒性作用的可能靶分子;
利用同源建模和分子对接技术,预测和模拟杂质与靶分子之间的相互作用。
完成了对C-7位含有氨噻肟结构的头孢菌素结构与神经毒性[184]、心脏毒性[185]的关系探讨,C-3位含有巯甲基四氮唑结构的头孢菌素结构与胚胎毒性[189]等的关系探讨;
关键词:杂质谱 化学药品 杂质鉴别 杂质评估 药品标准
分享至:
![]()
![]()




鄂公网安备 42011102004299号
© 2014-2024 前衍化学科技(武汉)有限公司 版权所有 鄂ICP备20009754号-1

