虽然中国境内基本完全控制了疫情,但是国外的情况总是让我觉得很揪心,世界是一个整体,国外的情况不好,中国的经济发展也会受到影响,希望大家都早点迈过这道坎,生活回归正常。
根据国外的疫情情况,按2020年6月28日的数据,国外有4129602为新冠感染者,且还在不断的增加,如果每为患者需要1克瑞德西韦制剂,需要4130公斤的瑞德西韦,目前API市场售价是1500一克,100万一公斤,这产品极具有研就价值,现在我就做了一点简单的分析和调研。我觉得对瑞得西韦的需求量还是很大的,我4个月前写了一篇有关瑞得西韦合成工艺的文章,一直犹豫是否要公开,犹豫了4个月,后来工艺又有了许多优化和改进,今天决定一起公开,希望大家转发分享,也希望疫情早点过去,言归正传,现在开始谈瑞德西韦。
瑞德西韦(Remdesivir,GS-5734)是一种在研的广谱抗病毒药物,属于RNA依赖的RNA聚合酶(RdRp)抑制剂,可通过抑制病毒核酸合成发挥抗病毒作用。目前,多项临床试验正在调查该分子治疗COVID-19的潜能。

该化合物的合成已经申请了多项世界范围的专利,药化方面的数据于2017年已经公开,发表在美国化学会ACS旗下著名的期刊J. Med. Chem.上(J. Med. Chem. 2017, 60, 1648−1661)
初期为了满足细胞水平的活性测试,需要快速的拿到筛选活性化合物,吉利德的科学家团队,以最简单粗暴的方式,设计了一条路线合成瑞得西韦,路线如下:
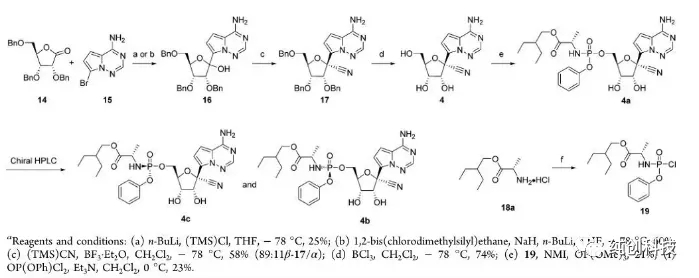
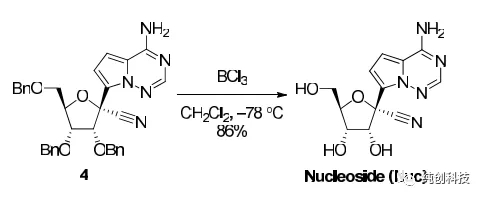
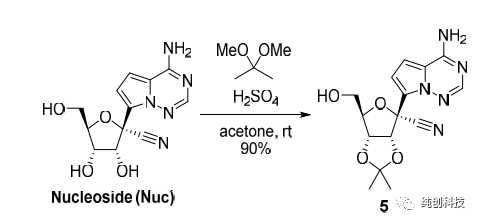
该路线首先将化合物15的氨基(-NH2)使用TMS保护,再在丁基锂的作用下与内脂14进行糖苷化反应然后将生成产物与化合物14反应再脱TMS保护生成化合物16(这里面有一个化合物15的芳香伯胺的原位硅保护和脱保护过程)再将化合物16进行氰基化引入氰基(-CN)得到17,化合物17在BCl3作用下脱苄基得到化合物4,4与化合物19反应得到消旋化合物4a,最后通过手性HPLC拆分离纯出4c与我们想要的终产物Remdesivir(4b)。
虽然该路线不适合放大生产,但是在做从成千上万个化合物中筛选活性好的明星分子,这样的合成方法屡见不鲜,百试不爽,总之策略就是快,效率高。
在做了细胞水平的活性测试以后,在细胞测试实验中表现极好的活性,瑞得西韦脱颖而出,为了满足药理,药代,动物实验等方面的活性测试,吉利德的科学家,针对化合物合成中的问题,要想做大,必须重新设计合成方法,通过大量的实验,他们有第一条路线进行了一些局部的优化和改进,合成路线如下:

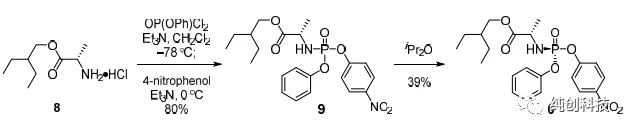
针对化合物筛选时合成路线的问题,吉利德的科学家通过大量的文献调研和实验,优化出手性合成的方法,实现了百克级别制瑞得西韦的合成。他们使用碘代物20替换第一代路线的溴代物15,并改变反应条件使这一步的产率由第一代的25%提升至40%,再通过反应条件的优化,找到了不对称氰基化的方法,改方法技巧性极强,合成设计极其巧妙、在顺利得到化合物4后,通过对化合物4邻位双羟基(-OH)的保护,高产率(90%)地得到了化合物21。
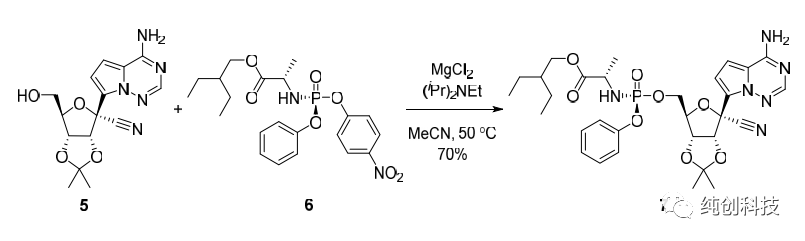
此条路线中,最关键的步骤是单一构型化合物22b的合成。化合物18a与苯基二氯磷酸酯及对硝基酚反应得到消旋化合物22a(产率80%),经过多种溶剂、多种条件的筛选探索,药物化学家发现将消旋化合物22a置于异丙醚中轻轻搅拌22h后,过滤后可得白色固体,此白色固体即为单一构型化合物22b,最重要的手性便这样引入了,化合物21与单一构型化合物22b反应(一定要高纯度),再脱保护,最终得到单一构型的目标化合物Remdesivir(4b)。这条路线可以合成百克级别的Remdesivir,用于后期研究中,但是问题很多,比如合成化合物16时,后处理需要使用柱层析,在合成几十公斤的化合物16时,五元内酯环活性低、易于烯醇化和消除、卤代碱基类似物脱卤等关键难题,需要使用很多的大层析柱,另外由于放大效应,反应比较杂,收率不太稳定,放大的时候仍然出现许多问题,曾经尝试过在这一步反应中上大位阻的保护基,收率可以提高到50%~60%左右,但是大家想想,为了做几十公斤的中间体,在车间里同时并列使用几十根大层析柱的场景,是比较头疼的事情,且不安全,效率低下。
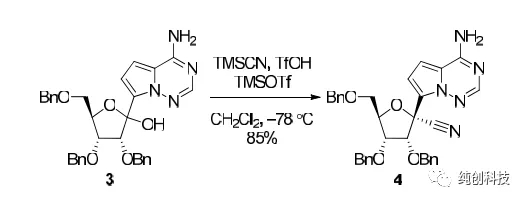
另外上氰基的时候,在酸性条件下加入如TMSCN,虽然是超低温反应,但是体系中有HCN产生,很不安全,还有在酸性环境下,产物有一些分解,产生了杂质,降低了产品的收率
为了建立安全,可靠的合成方法,我突然发现吉利德公司负责工艺的科学家又开发了新的工艺,他们尝试了一些新的工艺改进,方法如下:
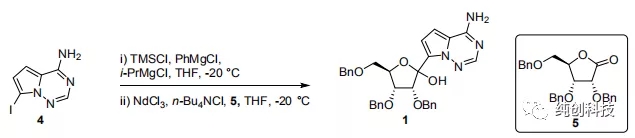
在反应釜中加入无水氯化钕(III)(1.0当量),正四丁基氯化铵(1.0当量)和四氢呋喃(THF,9体积),并将所得混合物在环境压力下在约66℃下浓缩至约4.5体积。加入THF(4.5体积),并重复浓缩两次。将混合物冷却至约20℃,化合物5(282kg,1.0当量)。约30分钟后,将混合物冷却至约-20℃并保存备用。
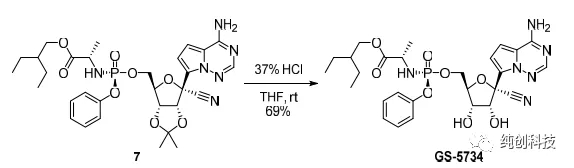
在另外一个反应容釜中,加入化合物4(1.10当量)和THF(5体积),并冷却至约0℃。缓慢加入三甲基氯硅烷(1.10当量),保温0℃30分钟后,将混合物冷却至约-10℃。缓慢滴加加苯基氯化镁的THF溶液(2M/L,2.17当量),并且在约30分钟之后,将反应混合物冷却至约-20℃。缓慢滴加异丙基氯化镁(2M的THF溶液,1.13当量),保温约2小时后,将格氏反应混合液转移至化合物5/ NdCl3 / n-Bu4NCl / THF混合物中。将所得混合物在约-20℃下搅拌,并且在约8小时后,添加乙酸(1体积)和水(4体积)的溶液。将所得混合物加热至约20℃,并加入乙酸异丙酯(i-PrOAc)。分离各层,有机层依次用碳酸氢钾水溶液和氯化钠水溶液洗涤。将有机层在真空下浓缩至约4.5体积,并加入i-PrOAc。将有机层用水洗涤两次,并在真空下浓缩至约4.5体积。加入i-PrOAc,并重复浓缩。将混合物过滤并将滤液在真空下浓缩至约3体积。加入甲基叔丁基醚(MTBE),并将体系降温至约20℃。加入化合物1的晶种,然后加入正庚烷,并将体系冷却至约0℃。通过过滤分离固体,并用MTBE /正庚烷混合液润洗滤饼。将得到的固体在真空下干燥,以69%的收率和100%的纯度得到化合物1。各种分析数据和先前报道的数据完全一致。
本方法使用了 NdCl3 / n-Bu4NCl /的复合盐调控亲核性,以69%的收率和100%的纯度得到化合物1,避开了柱层析,且做到了200过公斤的化合物1
另外就是上氰基的问题,1:是立体选择性的问题,大量化合物1 / TFA与TMSOTf / TMSCN结合使用所需的工作时间增加,这可能会对非对映选择性和收率产生负面影响,2:产生的HCN:,导致操作危险性很大,为了解决这两问题,吉利德的工艺科学家尝试了连续流的方法
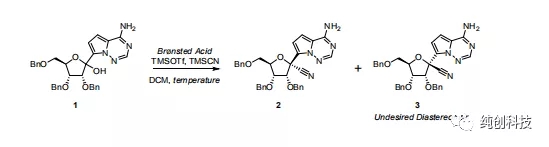
他们筛选了大量的条件,就我感觉,摸索工艺做毫克级别的反应,是一个非常粗糙的数据,但是还是介绍一下,数据如下:
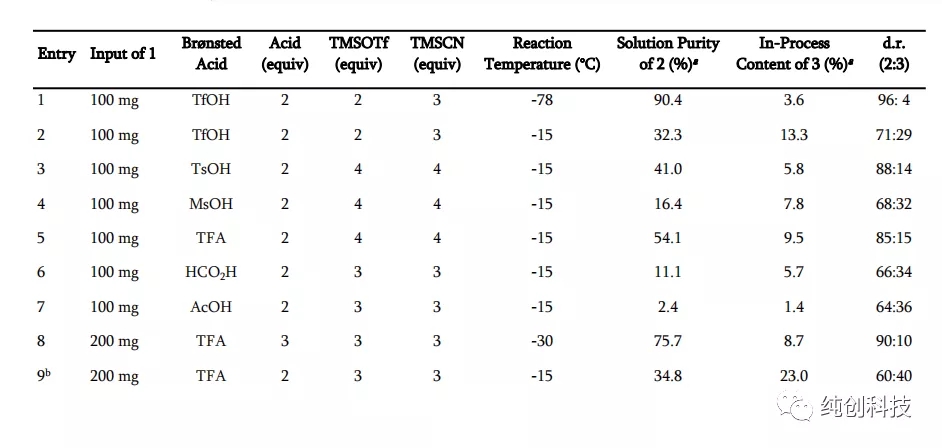

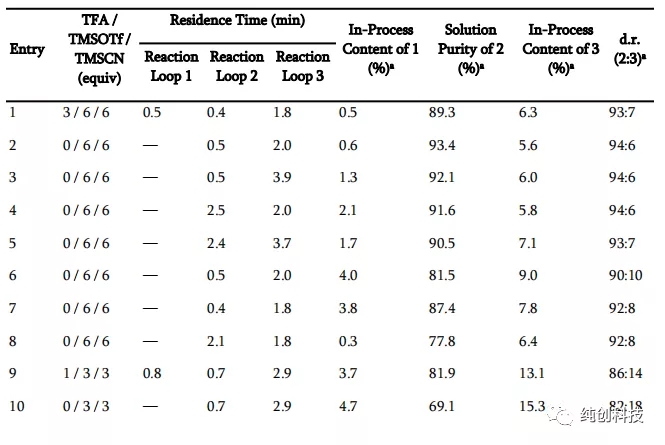
反应条件:将化合物1在DCM中的溶液冷却至设定的温度,并依次添加指定的Brønsted 酸,TMSOTf和TMSCN。a通过LC分析确定纯度。 b反应条件:将化合物1的DCM溶液冷却至设定温度,并添加TMSCN,然后加入TFA和TMSOTf。 c分别在大约30分钟内添加TMSOTf和TMSCN。 d反应条件:在-30°C下,将TFA添加至化合物1在DCM中的溶液中,然后添加预冷至-30°C的TMSCN和TMSOTf在DCM中的溶液。
首先观察温度升高的问题,从-78℃升高到-15℃,可以观察到化合物2含量在降低,说明产物转变成了杂质,将温度降低到-30℃,产物增加。
通过这个表格发现TFA和TfOH的优势相对明显,在使用TFA时,当温度升高到30°C时,TMSOTf和TMSCN的用量都增加到6当量时,反应的选择性增加,提供了93:7的非对映选择性,而在使用TfOH时,温度保持在-78 °C,TMSOTf和TMSCN的用量为2当量和3当量时,提供了96:4的高非对映选择性,加料顺序对非对映选择性也有影响,在添加TFA和TMSOTf之前添加TMSCN会减弱选择性。
这个表格总体设计不是科学,没有时间参数,温度低度变化幅度太大,不精细,另外使用TfOH时,数据太少了,需要补充,总之问题很多,估计是吉利德的科学家不想公开的太多吧。
结晶工艺优化:
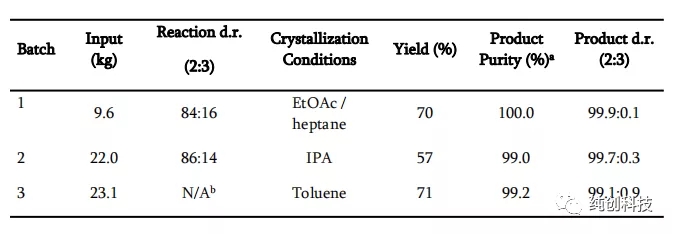
反应条件:将TFA(3当量)加入到-30°C的DCM中的化合物1的溶液中,然后将TMSCN(6当量)和TMSOTf(6当量)在DCM中的溶液预冷至-30°C 添加。通过LC分析确定纯度。 b未分析反应样品,以最大程度减少暴露于危险反应混合物中。
在一次性投料化合物1,9。6公斤,22公斤,23.1公斤时,从表格上看,在混合溶剂乙酸乙酯和庚烷中结晶效果最好,以及在单一溶剂甲苯和异丙醇中结晶次之。
连续流化学条件的优化
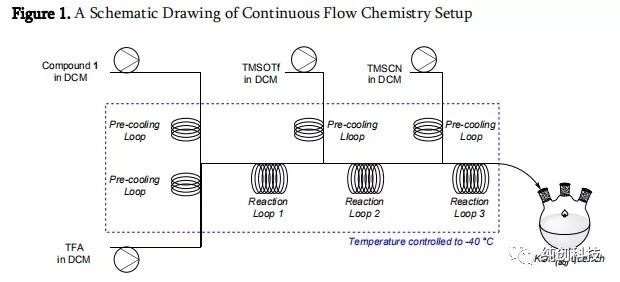
使用连续流反应器,每次投料1g,做了一些的的小试平行实验,我们发现
在TFA为3当量,TMSCN(6当量)和TMSOTf(6当量),整个体系预冷温度控制在-40°C,化合物1溶解于DCM中,通过盘管循环预冷,和第2个同样也通过盘管循环预冷的TFA和DCM的混合液混合,在第一个反应器混合盘管循环后,导入第3个通过盘管循环预冷的TMSOTf的DCM混合液,在第二个盘管反应器反应,接着导入第三个通过盘管循环预冷的TMSCN的DCM混合液,再在第三个盘管反应器反应中反应,后导入到含有KOH的接收瓶淬灭反应,控制了HCN的产生。
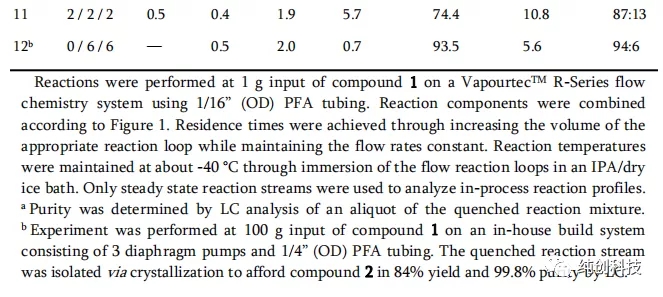
通过实验数据,发现一个结果和第二个结果比较理想,特别是第二个,居然不用加入TFA也获得了很好的结果,那为什么还要加入TFA呢,是不是有一些矛盾,为了验证这个结果,又进行了下面的实验进行验证。

使用SS连续流系统,对100克化合物1(不使用TFA)进行了实验,以确认系统稳定性。尽管起始原料的残留量略有增加(2.5%)以及非对映选择性的降低(91:9),但所得结果仍在预期之内(表4,第1项数据)。当时,这些低于预期的结果归因于实验开始和结束时的扩散,因为使用SS反应器系统进行此较小规模的试验运行时,收集到大量的非稳态反应混合物。在确认SS系统正常运行后,使用首选表3中条第12项数据表明的TFA为0当量,但是遇到了更为明显的挑战。观察到原料的不完全转化和非对映选择性的显着降低(表4,第2项数据)。使用先前使用的系统对设备的设置和执行进行调查(使用VapourtecTM系统进行小规模实验,以及使用内部3/16英寸PFA反应器系统或SS反应器系统进行100 g实验),并使用2.75 kg SS反应器系统中,当未向反应混合物中添加TFA时(表3的第11-12项数据和表4的第1项数据与表4的第2项数据相比),表明反应器的配置和结构材料不是改变结构的原因。反应性能。
寻找小规模试验与2.75千克试验运行之间差异的替代解释。假设残留的水可能通过TMSOTf的水解而产生微量的TfOH。然后,生成的TfOH可以充当催化剂,同时催化转化的选择性和转化率。这种作用可能会对较小规模的反应(即1克甚至100克)产生更大的影响,而随着规模的增加(即2.75千克)而影响减小。为了验证该假设,使用SS反应器系统将TfOH引入到150 g反应的TMSOTf / DCM储备溶液中,发现转化率和非对映选择性已完全恢复(表4,第3项数据)。进一步评估了TFA在反应混合物中的使用,并使用SS反应器系统进行了150 g规模的实验(表4,第4~6项数据)。发现在工艺过程中0.5摩尔当量的TFA,得到可接受的转化率和选择性(表4,第5项数据)。为了适应三泵系统流量中的潜在波动,并提高过程的稳定性,在后续运行中选择了1.0当量的TFA。在实施此更改过程中,TFA / TMSOTf进料管线的背压增加了, 最终堵塞。在进行其他实验以评估堵塞的原因后,发现TFA(熔点为-15.4°C)在-40°C的温度下在TMSOTf / DCM混合物中的溶解度不好,并且可能在进料的预冷却过程中析出结晶。为了避免此问题,工艺中已去掉了TFA / TMSOTf / DCM进料预冷却回路。此外,将反应温度从-40°C调节到-30°C,以降低TFA从TFA / TMSOTf / DCM进料溶液中结晶的风险。这些变化对反应的性能没有实质性影响,因为化合物2的溶液纯度(和反应的非对映选择性)是相似的,尽管反应温度升高并且TFA / TMSOTf / DCM预冷回路已移除( 表4,第5,6项数据)。
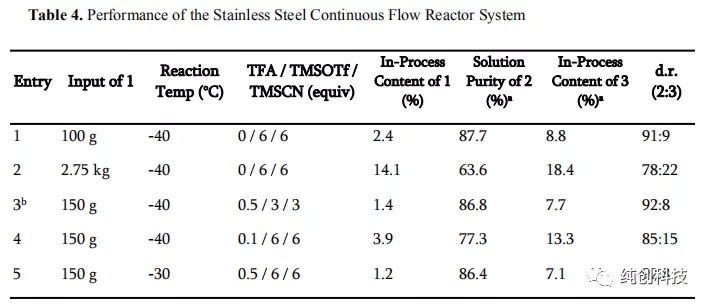

在这些新的条件下,随后的5次投料2.75 kg的化合物1进行实验验证,提供了高溶液纯度和非对映选择性表5,第2~6项数据表明TFA是有利于反应收率的提高。合并来自多个批次的猝灭的流出物,并以66%的收率和99.7%的纯度分离出13.0kg的化合物2。

在工厂使用连续流化学法合成化合物2
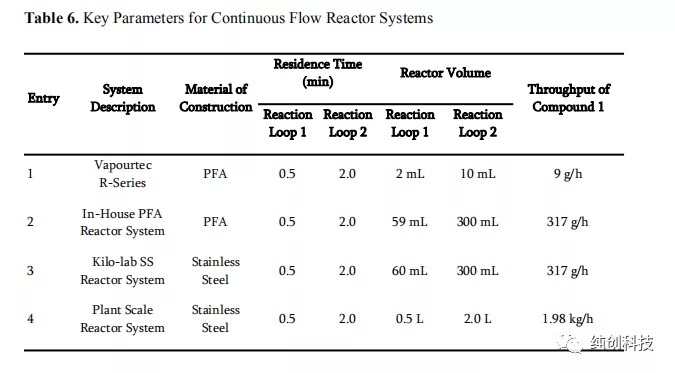
使用各种连续流反应器系统进行克级别到千克级别的结果,并将应用于每个系统的工艺参数用于设计工厂连续流反应器系统的基础。以下主要因素用于工厂系统参考。 首先,必须妥善控制生产体系,以确保安全处理含氰化物的反应混合物。为了解决这个问题,流动反应器和辅助设备均由不锈钢制成,并在使用前进行了严格的压力测试。 其次,生产体系需要具有在合理的时间内生产数百公斤化合物2的能力。表6列出了不同系统的工艺参数的比较。可以清楚地观察到停留时间,反应器体积和产量(化合物1的消耗量)之间的相互作用。从该表可以看出,容积为2.5 L的不锈钢管反应器,可以实现约2 kg / h的通过量,从而能够在约4天的时间内处理200 kg的化合物1。对于生产要求,该连续生产时间是可以接受的。为了始终如一地达到三种进料中每种进料的期望流速,选择了三个由不锈钢和PTFE制成的隔膜泵,它们配有背压调节器作为进料装置,并在使用前进行了校准。 最后,生产体系需要具有足够的冷却能力和效率,以便在反应之前将试剂溶液调节到约-30°C,以及迅控制反应过程中产生的热量,确保生成高质量的产品连续流生产。这些因素通过使用SS结构和管道式反应器系统控制,实现快速热量传递,并使用大容量加热/冷却模块。
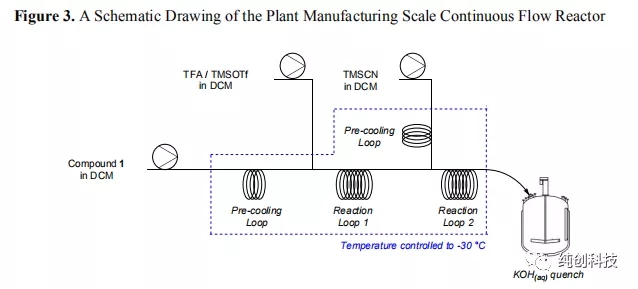
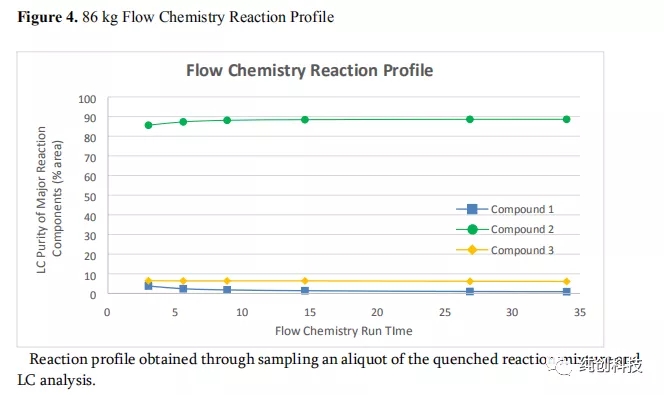
在工厂大规模的连续流反应器系统的安装和调试完成之后,在化合物1的投料量为86 kg的条件下进行了试验运行,以验证工厂的可操作性,用于合成化合物2的系统。如图4所示,在短暂的诱导期后,即获得系统的稳定操作,在整个流动反应过程中观察到一致的反应曲线。数值结果可见于表7(第1项数据),通过表7的第1项数据和表5的第2-6项数据相比,表明与放大生产实验系统相比,SS公斤级的系统提供了与化合物2相似的反应非对映选择性和产品纯度。用公斤级实验批次相同的方式将淬灭的反应混合物进行水后处理,用于化合物2在DCM中在溶液储存。在将溶剂置换成甲苯后,通过重结晶后过滤,获得68kg的化合物2(表7,实验数据1)。改工艺生产的瑞得西韦可满足临床的需要。
随后使用相同的工艺,进行了另外两次生产,投料量分别为280 kg和250 kg,分别得到204 kg和198 kg化合物2(表7,第2,3项数据)。

化合物2的制备


1:普通工艺:
投料化合物1(23 kg,1.0当量)在DCM(10体积)中的溶液中冷却至-40°C,加入三氟乙酸(3.0当量), 加入TMSOTf(6.0当量)和TMSCN(6.0当量)在DCM(5体积)中的预冷溶液(-30°C),同时确保反应釜体系内部温度低于约-25°C。加毕, 将反应混合物在低于-30℃下搅拌不少于10分钟后 ,取样,通过HPLC监测反应完全,用预冷的KOH(35.8当量,14倍体积的水,预冷到-10℃)溶液淬灭反应。
将两相混合物升温至室温。分离有机层,并用氯化钠水溶液洗涤3次。有机相在真空下浓缩至约4体积,用DCM(8体积)稀释并在真空下浓缩至约6倍体积。将混合物过滤,滤液浓缩至约4倍体积。将浓缩物用甲苯(19体积)稀释,并在约50℃下真空浓缩至约14体积。将混合物加热至约90℃,冷却至约55℃,加入化合物2的晶种。将混合物在约55℃下搅拌养晶约1小时,并在约6小时内缓慢降温至约0℃。过滤得到固体,用甲苯(3体积)洗涤滤饼。将固体在约50℃下真空干燥,以71%的收率和99.2%的纯度提供化合物2。
2:连续流化学过程生成化合物2:
化合物1(250 kg,1.0当量)在DCM(15.0体积)(进料1),TMSOTf(6.0当量)和TFA(1.0当量)在DCM(4.4体积)中的储存溶液(2) 在分开的反应器或进料容器中制备进料2)和DCM(4.5体积)中的TMSCN(6.0当量)(进料3)。将进料1以约504 mL / min的流速用计量泵输送通过约-30°C的预冷却回路,并将进料2以约207 mL / min的流速泵送。将进料1和2在约-30℃的盘管反应器1中合并约30秒。然后将流出物与进料3(通过约-30°C的预冷却回路以约189 mL / min的速度泵输送)在约-30°C的盘管反应器2中混合约2分钟。合并的出料口的流出物,直接收集到装有KOH(19.7当量)在水(8体积)中的溶液的容器中,保温10°C。 将混合液升温至约20℃,然后加入2-丙醇并分离各层。有机层用氯化钠水溶液洗涤两次,过滤,浓缩,所得溶液,加入甲苯到滤液中并将混合物浓缩。将混合物加热至约55℃,然后冷却至约0℃,过滤所得浆液,用甲苯冲洗并在约60℃下干燥,得到 化合物2,收率为78%,纯度为99.9%。分析数据与文献中先前报道的数据一致。监测氰化过程(实验室规模和工厂规模)的废料中氰化物含量,并用次氯酸钠进行处理,直到氰化物含量低于氰化物试纸的检测极限为止。


 .
.







API Remdesivir

换一个结晶条件,过滤时,API Remdesivir在布氏漏斗中的外观和形态



Remdesivir的制剂
化合物Remdesivir在水性介质中相对不溶且化学上不稳定。在pH 约4至5之间的溶液中最稳定。在pH为4或以下时,化合物的溶解度增加。
 .
.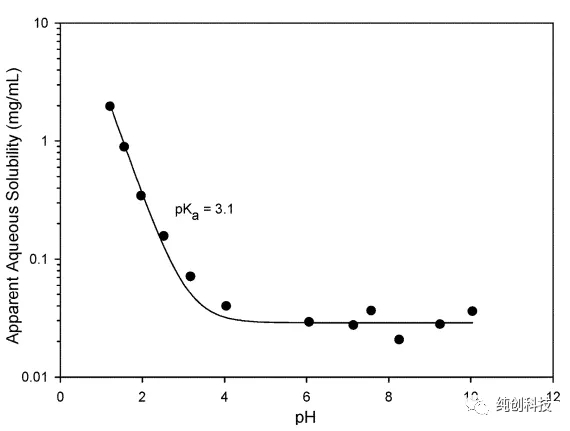
(Remdesivir在不同pH时的稳定性)
注射用Remdesivir冻干制剂是一种不含防腐剂的白色至灰白色或黄色冻干固体。除活性成分外,冻干制剂还包含以下非活性成分:注射用水,磺丁基倍他环糊精(SBECD)和盐酸和/或氢氧化钠。SBECD是阴离子型高水溶性环糊精衍生物,能很好地与药物分子包合形成非共价复合物,从而提高药物的稳定性、水溶性、、安全性、降低肾毒性等。

冻干组合物制备方法:
将环糊精溶解在水中,向溶剂中加入酸调节pH小于2,将化合物Remdesivir加入到该酸化的混合物中形成复合物的悬浮液,向溶液中加入碱调整pH至约3.5,形成过饱和溶液。根据需要向其中加入水调节化合物的浓度,并将混合物冻干。
关键词:新冠病毒 特效药 瑞德西韦 合成工艺
分享至:
![]()
![]()




鄂公网安备 42011102004299号
© 2014-2025 前衍化学科技(武汉)有限公司 版权所有 鄂ICP备20009754号-1






 发询盘
发询盘