 资讯
资讯
纳米材料与类器官结合产生的机遇
类器官是一种来源于自体细胞培养的多细胞三维组织,被认为是动物模型的优质替代模型。类器官模型具有比二维细胞模型寿命更长以及完全没有与动物试验相关的伦理问题等优势。此外,类器官具有器官的鲜明特征,与实际器官具有高度的相似性,类器官的显著细胞异质性复制了真实器官的结构和复杂性。 类器官作为利用干细胞自组织形成的三维微型器官模型,近年来在基础研究和个性化医疗中展现出巨大的潜力。这些微型结构不仅能够模拟原生器官的结构和功能,还为疾病模型、药物筛选和再生医学提供了全新途径。与此同时,纳米技术的迅速发展为类器官的构建与应用注入了新的活力。 类器官与纳米材料的结合:新型工具的诞生 类器官的构建依赖于干细胞的分化和自组织能力,而纳米材料通过物理信号和化学信号的精准调控,显著提升了干细胞的分化效率。例如,金纳米颗粒(AuNPs)能够作为纳米载体,将神经营养因子递送至诱导多能干细胞(iPSCs)形成脑类器官,显著提高相关基因的表达水平。此外,磁性纳米颗粒(MNPs)通过动态磁场作用,成功实现了胚胎干细胞(ESCs)向心肌细胞的定向分化。 纳米材料还在构建3D微环境方面展现了独特优势。传统的类器官培养主要依赖二维(2D)细胞培养技术,这种技术难以完全模拟体内的三维微环境。纳米材料,如氧化锌(ZnO)纳米棒和石墨烯氧化物(GO),能够提供三维支架结构,改善细胞与细胞外基质(ECM)之间的相互作用,促进类器官的成熟和功能化。通过磁悬浮培养技术,将含有磁性纳米颗粒的细胞悬浮于液气界面,为类器官的构建提供了高效且无支架的解决方案。 纳米材料助力3D打印类器官 3D打印技术为类器官的规模化生产和精准构建提供了新工具,而纳米材料的引入进一步提升了打印效率和生物功能性。例如,甲基丙烯酸改性壳聚糖(ChMA)与纳米羟基磷灰石(nano-HAp)复合的生物墨水,不仅在打印过程中表现出良好的流变性和结构稳定性,还能显著促进骨髓间充质干细胞(BMSCs)的增殖与分化。另一项研究利用含超顺磁性氧化铁(USPIO)纳米颗粒的凝胶打印胆管类器官,不仅能够实时监测打印结构的功能状态,还可通过磁共振成像(MRI)追踪组织修复进程。 尽管如此,3D打印类器官的稳定性和可重复性仍然是制约其广泛应用的重要因素。研究者通过将金属纳米颗粒与水凝胶基质结合,改善了打印过程中材料的机械性能和生物相容性,为未来大规模类器官的定制化生产铺平了道路。 纳米材料在毒性评估中的应用 纳米材料的生物安全性是其能否广泛应用于类器官构建的重要前提。类器官由于其高度模拟体内组织环境的特性,为纳米材料的毒性评估提供了理想平台。例如,利用肾类器官评估黑磷量子点(BP-QDs)的生物安全性发现,这些纳米材料可通过非折叠蛋白反应导致肾脏损伤。此外,通过脑类器官研究发现,较高浓度的银纳米颗粒(AgNPs)会显著抑制神经突起的生长,并引发神经发育毒性。这些研究不仅揭示了纳米材料潜在的毒性机制,也为其安全性优化提供了科学依据。 纳米材料与类器官的结合为个性化医疗带来了革命性机遇。从毒性评估到疾病模型,从3D打印到功能化设计,纳米技术正在推动类器官从实验室走向临床。然而,为了实现这一愿景,研究者需进一步解决纳米材料的安全性、可重复性及其在类器官中的长期稳定性等问题。随着技术的不断进步,纳米材料与类器官的交叉研究必将在生物医学领域开辟更加广阔的天地,为患者提供更精准、更高效的个性化治疗方案。 前衍可供研究的部分纳米材料 中文名 CAS号 链接 纳米氧化锌 1314-13-2 订购 复壁碳纳米管 308068-56-6 订购 纳米碳酸钙 471-34-1 订购 石墨烯 1034343-98-0 订购 多壁碳纳米管 1333-86-4 订购 纳米级氧化钛 1317-80-2 订购 纳米二氧化钛 13463-67-7 订购 磷酸锆(纳米) 13772-29-7 订购 黑磷纳米片 7723-14-0 订购 碲化锡 12040-02-7 订购 纳米二氧化硅 60676-86-0 订购 亲水型纳米二氧化硅 7631-86-9 订购 疏水型纳米二氧化硅 68611-44-9 订购 纳米碳化硅 409-21-2 订购 巴克明斯特富勒烯 99685-96-8 订购 纳米金刚石 7782-40-3 订购 富勒烯 131159-39-2 订购 纳米四氧化三铁 1317-61-9 订购 纳米氧化钙 1305-78-8 订购 纳米氧化钴 1308-06-1 订购
2025-02-28
环保新希望-光催化PFAS低温脱氟技术取得重大进展
全氟和多氟烷基物质(PFAS)具有独特的热稳定性、化学稳定性、疏水及疏油特性等,广泛应用于化工、电子、医疗设备、纺织机械、核工业等领域。然而碳-氟键的惰性导致PFAS在自然环境或温和条件下难以降解,PFASs也因此被称为“永久化学品”。例如,大量废弃的全氟辛酸(PFOA),全氟辛磺酸(PFOS)等在自然界无法降解,已在自然水域、生物体甚至人体内检测到。它们在人体的富集会影响发育、生殖等,还会造成肝中毒、内分泌紊乱及致癌等危害。此外,特氟龙在260摄氏度可维持多年不分解,在500℃以上分解时则会释放出有毒气体。因此,被废弃于自然界的PFAS引发了一系列环境及健康问题。 近日,中国科学技术大学合肥微尺度物质科学国家研究中心及化学系的研究团队创制了一种超级光还原剂,并基于此发展了低温的催化还原特氟龙等全氟及多氟烷基化合物的高效降解。相关成果已以“Photocatalytic low-temperature defluorination of PFASs”为题发表在Nature上。 目前,全氟和多氟烷基物质(PFAS)的降解方法主要包括水热法、机械化学法、电化学法、等离子体法以及过渡金属催化法等。现在的降解方式主要还是通过高温高压或输入大量能量对全氟和多氟烷基物质的结构进行破坏。这些方法具有能量损耗大、成本高、处理不完全、毒性大、污染高等缺点。亟需寻找一种廉价、原子经济性高、绿色环保、温和的方式来降解这些全氟和多氟烷基物质(PFAS),尤其是PTFE的降解。 围绕上述挑战,研究团队基于在特定光照下具有超强还原性的原理,设计创制了超级有机光还原剂(取名为KQGZ),首次实现了低温下(40-60 ℃)的特氟龙及小分子PFAS的完全脱氟矿化,将其高效回收为无机氟盐和碳资源。 光催化PFASs低温脱氟示意图 还原剂是能够提供电子的化学物质;而超级还原剂则是能够把电子注入到还原电位低于负 3 伏特的化学键的电子供体。 超级光还原剂KQGZ的设计灵感源于高度扭曲的咔唑核结构,这一结构能够有效促进电子的导出和还原反应。这一发现不仅为PFAS的降解提供了新的思路,也推动了光还原催化剂的研究进展。该研究首次证明,激发态氧化电位并非判断光催化剂还原能力的唯一标准,超级还原剂的创新性设计使其在现有的百余类反应中均取得了令人满意的结果。 KQGZ的广谱性能使它能够破坏强碳-杂原子以及杂-杂原子键,使得在环境中难以降解的PFAS能够得到彻底去除。这种技术的突破为解决持久性有机污染物问题提供了新的解决方案,具有极大的环保意义。 前衍可供研究的部分PFAS(全氟和多氟烷基物质) 中文名 缩写 CAS号 链接 全氟戊酸 PFPeA 2706-90-3 订购 全氟己酸 PFHxA 307-24-4 订购 全氟庚酸 PFHpA 375-85-9 订购 全氟辛酸 PFOA 335-67-1 订购 全氟壬酸 PFNA 375-95-1 订购 全氟癸酸 PFDA 335-76-2 订购 全氟十一酸 PFUnDA 2058-94-8 订购 全氟十二酸 PFDoDA 307-55-1 订购 全氟丁烷磺酸 PFBS 375-73-5 订购 全氟己烷磺酸 PFHxS 355-46-4 订购 全氟辛烷磺酸 PFOS 1763-23-1 订购 全氟癸烷磺酸 PFDS 335-77-3 订购
2025-02-21
人工智能推动科研范式第五次变革
科研范式”这一概念第一次被提出,是在美国科学哲学家托马斯·库恩所写的《科学革命的结构》一书中。他认为,“范式”是从事某一科学的研究者群体所共同遵从的世界观和行为方式,是为了保障科研活动高效有序运转所遵循的普遍法则。 在科学发展的特定时期,通常由一种范式主导。随着主导范式被广泛接受和长期应用,往往会出现当前范式难以解释的科学发现。当这些问题逐渐积累到引发质变时,新的科研范式呼之欲出,并逐渐取代旧范式。科学的发展就是通过一系列“范式转换”实现,每一次转换都是一次科学观念的根本变革。 过去千百年来,传统范式经历了四个阶段的演化历程:第一阶段为经验范式,主要通过实验描述自然现象;第二阶段为理论范式,主要通过模型或归纳法进行科学研究;第三阶段为计算范式,指使用计算机进行仿真模拟来解决各个学科中的问题;第四阶段为数据范式,指通过大数据分析研究事物内在的关系得到结论。 现今科学研究中,尤其是材料科学、合成生物学、化学、天文学和地球科学等领域,科学数据呈现出爆炸式增长。为了从这些海量数据中挖掘出知识规律,传统方法(如计算机仿真和手动实验)常显得力不从心。例如,在2005至2015年间,基因组学的序列数据几乎每7个月就要翻一番。而在天文学中,自1990年起运行的哈勃太空望远镜每周能传回约20GB的原始数据。 这正是科研人员长期以来面临的难题:一是科研成果在实际应用中的挑战;二是数据收集、处理与分析效率较低;三是大部分科研团队依旧采取“作坊式”工作模式,而平台化合作较为稀少;四是在材料研发等领域依赖经验和试错的方式进行突破。这些庞大的数据需要进行分类、回归、聚类、关联分析、时间序列分析以及异常检测等处理。只有在完成这些步骤后,隐藏的模式和未知的相关性才会浮现,否则只是无效冗余。同时,现代科学已进入复杂体系时代,传统的计算方法难以应对越来越多变量和计算复杂度所带来的瓶颈。 在此背景下,人工智能的核心技术——深度学习展现出独特的优势。深度学习的设计本就源自对大数据的需求,数据处理不仅是它的强项,也是其生存和发展的基础。深度学习能在大量数据中找出规律,减轻数据爆炸带来的挑战。例如,人工实验员一天难以完成的重复实验,通过自动化平台在一天内便可高效完成上百次,大幅提高实验数据的准确性和一致性,而高质量的实验数据正是模拟和训练的基础。 人工智能技术的发展使科学家开始超越传统的四大科研范式,依托先进的计算技术,推动了第五代科研范式——利用人工智能技术对自然现象进行学习、模拟、预测和优化,从而推动科学发现和技术创新。相比于传统的科研方法,这种科研范式不仅显著提升了科学问题的解决效率,还为科研人员提供了新的研究角度与方向,开辟了探索未知的全新路径。 一个典型的例子是,2024年度诺贝尔物理学奖与化学奖均与人工智能研究相关。这一方面肯定了人工智能在促进基础科学进展中的关键作用,另一方面也预示着物理、化学等传统学科将变得更加开放,科学家们不再局限于传统的“可解释性”研究模式,而是通过实验校准不断完善模型,从而获得更全面的理解。 尽管人工智能带来了诸多益处,其应用仍需保持谨慎。科学界对其介入依然持谨慎态度,不同的声音从未停止发出。例如在生物学研究中,作为研究对象的人类个体信息和医学特征信息都包含了较多的隐私内容。在数据挖掘和分析过程中,如果数据隐私得不到有效保护,在一定程度上会影响生物学的发展以及科学研究的可信度。虽然已有部分专家学者提出了一些创新技术手段,在保证数据安全的情况下进行数据共享交换以及模型的搭建训练,但这一问题仍需进一步探索与解决。
2025-02-14
七起危险化学品违法案例
上海市公安局浦东分局破获周某仙等11人非法经营危险化学品案 2023年5月25日,浦东分局破获一起非法经营危险化学品案,累计抓获涉案人员周某仙(女,51岁,浙江衢县人)、陈某锋(男,43岁,河南商城县人)等11人,查获工业甲醇等危险化学品20余吨。 经查,周某仙、陈某锋在未取得危险化学品经营许可证的情况下,通过非法手段从江苏太仓等地采购大量工业甲醇,储存在浦东区私建的2处仓库内;招揽9名分销人员通过社交平台等方式向饭店兜售、运送工业甲醇,冒充“环保油”从中非法获利。 犯罪嫌疑人周某仙、陈某锋等11人行为均触犯了《中华人民共和国刑法》第二百二十五条的规定,涉嫌非法经营罪;依据《中华人民共和国刑事诉讼法》第八十二条的规定,依法对犯罪嫌疑人周某仙、陈某锋等11人先行拘留,取保7人,决定起诉4人。 湖南省株洲市市场监管局查处株洲市星空化玻有限责任公司无证生产危险化学品案 2023年2月7日,株洲市市场监管局收到湖南省市场监管局产品质量违法线索交办函,对相关线索进行立案调查。 经查,2022年12月,株洲市星空化玻有限责任公司从湖南某有限公司购进浓度98%硝酸20000公斤。该公司在未取得工业级硝酸《全国工业产品生产许可证》情况下,生产浓度65%—68%(±2)的工业级硝酸5500公斤。上述违法行为存续期间,未对外销售,无违法所得,总货值金额14874元。当事人已停止生产该产品。 当事人的行为违反了《中华人民共和国工业产品生产许可证管理条例》第五条的规定,2023年4月21日,依据《中华人民共和国工业产品生产许可证管理条例》第四十五条的规定,株洲市市场监管局对当事人未取得工业产品生产许可证擅自生产列入目录产品的行为处以没收违法生产的浓度为65%—68%(±2)工业级硝酸5500kg、罚款人民币14874元的行政处罚。 广东省珠海市应急管理局严惩非法经营危险化学品违法行为案例 2024年3月15日,珠海市香洲区应急管理局执法人员检查发现珠海某贸易有限公司在位于金鸡路433号6栋副楼储存了氢氟酸、次氯酸钠等危险化学品共约20余吨。经查实,该公司从事次氯酸钠储存、稀释、分装、销售行为是未经许可的危险化学品经营行为。该公司经营氢氟酸等9种危险化学品未在公司的危险化学品经营许可证许可范围内,认定该公司未经许可经营危险化学品。该公司储存酒精、石油醚、氢氟酸,但未按照酒精、石油醚、氢氟酸的危险特性采取防火、防爆、防泄漏等安全防范措施,没有将危险化学品储存在危险化学品专用仓库内。该公司向香洲区应急管理局提交危险化学品经营许可证办证申请中的经营方式为贸易经营,但实际上该公司在申请的经营场所外租用场地长期从事带储存经营危险化学品活动。 9月12日,香洲区应急管理局依据《危险化学品安全管理条例》第七十七条第三款、第八十条第(四)项、《危险化学品经营许可证管理办法》第二十六条、《安全生产违法行为行政处罚办法》第五十三条的规定,对该公司违法行为作出处罚决定,合并处罚款人民币210,000元,没收违法所得人民币1,013,688.37元,没收违法经营的氢氟酸1吨、次氯酸钠溶液19吨,撤销危险化学品经营许可证。同时结合调查掌握的事实,根据国务院《行政执法机关移送涉嫌犯罪案件的规定》,拟将该企业法定代表人涉嫌构成危险作业罪一案移送公安机关。 广东省东莞市应急管理局查处危险化学品违法承租储存案 2024年8月8日,东莞市应急管理局麻涌分局(以下简称“麻涌应急管理分局”)执法人员对位于东莞市麻涌镇的东莞某仓储有限公司进行执法检查,在该公司内发现江门市某化工原料有限公司涉嫌存在违反《危险化学品安全管理条例》有关规定,未将危险化学品(乙醇胺、二乙醇胺)储存在专用仓库内。结合相关调查取证,发现该仓储有限公司将麻涌公司仓库出租给广州市某化工贸易有限公司,未与承租单位签订专门的安全生产管理协议,且未在《仓储服务合同》中约定各自的安全生产管理职责。 麻涌应急管理分局执法人员在案件调查的过程中依法对案件涉及单位的相关人员进行询问,并制作《调查询问笔录》,确认东莞某仓储有限公司把仓库出租给广州市某化工贸易有限公司后,未与承租单位签订专门的安全管理协议,且未在《仓储服务合同》中约定各自的安全生产管理职责。 涉事化工原料有限公司存在未将危险化学品(乙醇胺、二乙醇胺)储存在专用仓库内的行为,违反了《危险化学品安全管理条例》第二十四条第一款的规定,依据《危险化学品安全管理条例》第八十条第一款第(四)项的规定,决定给予处罚款人民币85000元的行政处罚。 涉事仓储有限公司存在将生产经营场所出租给其他单位,未与承租单位签订专门的安全生产管理协议,且未在租赁合同中约定各自的安全生产管理职责的违法行为,违反了《中华人民共和国安全生产法》第四十九条第二款的规定,依据《中华人民共和国安全生产法》第一百零三条第二款及参照《东莞市应急管理局适用〈中华人民共和国安全生产法〉行政处罚自由裁量标准(试行)》第33条第(1)项的规定,东莞市应急管理局麻涌分局决定就上述违法行为对该企业作出罚款人民币10000元的行政处罚、对该公司主要负责人黄某作出罚款人民币2000元的行政处罚。 安徽省安庆潜山市应急管理局严厉打击违法储存危化品案(三起) 案例1 2024年6月26日,安庆潜山市应急管理局执法检查过程中发现,叶某某在个人租赁的位于潜山市梅城镇某村民组的民房内,存放醇基燃料(主要成分为甲醇)约350千克。根据现场检查情况,通过查阅资料、谈话询问、抽样送检等,并经应急管理部危化品登记中心检测,叶某某储存的醇基燃料闭杯闪点为11.5℃,属于危险化学品。 该行为违反了《危险化学品安全管理条例》第二十四条第二款的规定,依据《危险化学品安全管理条例》第六条第二项,和《中华人民共和国安全生产法》第六十九条的规定,潜山市应急管理局于7月24日将该案件移送潜山市公安局进一步审理。 案例2 2024年7月12日,执法检查过程中发现潜山市某新能源科技有限公司将800余千克未知液体(主要存放在塑料吨桶)存放于痘姆乡一废弃小学内,该公司未取得相应资质。经第三方技术鉴定,该公司存放于原料桶3、原料桶6、原料桶7内的液体物质均属于危险化学品。 该行为违反了《危险化学品安全管理条例》第二十四条第二款的规定,依据《危险化学品安全管理条例》第六条第二项,和《中华人民共和国安全生产法》第六十九条的规定,潜山市应急管理局于9月14日将该案件移送潜山市公安局进一步审理。 案例3 2024年8月9日,高新区安监局对园区某危险化学品企业进行安全专项检查时,发现企业三号丙类仓库存放丙烯酸树脂,共24桶,每桶200kg,合计4800kg。丙烯酸树脂未按设计储存在甲类仓库,且设计最大储存量800吨,企业违规储存危险化学品。 该行为违反了《危险化学品安全管理条例》第二十四条第二款的规定,依据《危险化学品安全管理条例》第八十条第五项的规定,安庆高新区安监局对企业给予责令改正,处5万元罚款的行政处罚。
2025-02-08
2024年国内批准的新药,小分子仍占半数
2024 年中国 NMPA 共批准 90 款新药(据 Insight 数据库统计),其中国产有 37 款,进口 53 款。 在这些新药中,小分子仍是创新主力,其次是抗体类药物,其他类型药物包括多肽、ADC、细胞治疗、核酸等。 从疾病领域来看,肿瘤仍是创新药汇聚之地,2024 年共批准 31 款抗肿瘤新药;紧随其后的是罕见病和内分泌代谢领域,均批准 12 款新药;其他领域包括神经/精神、自免、感染、血液等,也都有创新产品获批上市。 值得指出的是,2024 年 NMPA 批准的 90 款新药中,已经有 19 款通过医保谈判纳入了 2024 年国家医保目录,包括 13 款国产新药和 6 款进口新药。 EGFR-TKI 继续内卷 肺癌领域 EGFR-TKI 药物在 2024 年持续发力,共有 3 款新药上市。 其中倍而达药业瑞齐替尼、圣和药业瑞厄替尼的获批上市,标志着国内三代 EGFR-TKI 药物市场已然形成六雄争霸局面,竞争持续白热化。 另外 1 款获批的 EGFR-TKI 新药是晨泰医药从阿斯利康引进的佐利替尼,与众不同的是,该药物具有完全穿透血脑屏障的能力,是全球首个针对肺癌脑转移的 EGFR-TKI 新药,为 EGFR 突变肺癌脑转移患者带来了新的治疗选择。 目前国内已获批的三代 EGFR-TKI 药物名称 企业名称 国内首次获批时间 具体适应症 奥希替尼 阿斯利康 2017.03 二线治疗EGFR T790M阳性NSCLC, 一线治疗EGFR突变NSCLC, EGFR敏感突变NSCLC的术后辅助治疗, 联合化疗一线治疗EGFR突变NSCLC 阿美替尼 翰森制药 2020.03 二线治疗EGFR T790M阳性NSCLC, 一线治疗EGFR 19del或21 L858R突变NSCLC 伏美替尼 艾力斯医药 2021.03 二线治疗EGFR T790M阳性NSCLC, 一线治疗EGFR 19del或21 L858R突变NSCLC 贝福替尼 贝达药业 2023.05 二线治疗EGFR T790M阳性NSCLC, 一线治疗 EGFR 19del或21 L858R突变NSCLC 瑞齐替尼 倍而达药业/石药集团 2024.05 二线治疗EGFR T790M 多个靶点迎来首款国产新药 首个国产 KRAS G12C 抑制剂 劲方医药自主研发的氟泽雷塞,是国内首个 KRAS G12C 抑制剂,信达拥有中国的开发商业化权益,24 年 8 月获批用于治疗至少接受过一种系统性治疗的 KRAS G12C 突变型晚期非小细胞肺癌。 KRAS 曾有「不可成药」靶点之称,随着 21 年安进公司的 Sotorasib 的上市,众多企业开始扎堆布局 KRAS,截止目前美国和中国各批准 2 款 KRAS G12C 抑制剂,均通过加速批准上市。 首个国产 TROP2 ADC 芦康沙妥珠单抗是科伦博泰开发的一款 TROP2 ADC 药物,已获批用于治疗既往至少接受过 2 种系统治疗的不可切除的局部晚期或转移性三阴性乳腺癌,是国内首个、全球第二个获批上市的 TROP2 ADC 药物。 此外,该药物还斩获了四项 CDE 突破性疗法认定,其用于三线和二线治疗晚期或转移性 EGFR 突变非小细胞肺癌的适应症,也分别在去年 8 月和 10 月申报上市,据 Insight 数据库预测有望在 25 年 Q1 和 Q2 获批。 首个国产 MEK 抑制剂 科州制药的妥拉美替尼获 NMPA 附条件批准上市,用于含抗 PD-1/PD-L1 治疗失败的 NRAS 突变的晚期黑色素瘤患者,成为首款国产 MEK 抑制剂。 淋巴瘤领域全球首个高选择性 JAK1 抑制剂 戈利昔替尼是迪哲医药自主研发的高选择性 JAK1 抑制剂,用于治疗复发难治性外周 T 细胞淋巴瘤(PTCL)。 PTCL 是一种高度异质性和强侵袭性的非霍奇金淋巴瘤,发病机制复杂,一线治疗方案以蒽环类化疗为主,一线治疗后复发患者预后差且缺乏更多有效治疗方案,戈利昔替尼的上市成为了全球首个通过靶向 JAK/STAT 通路治疗复发难治性 PTCL 的全新作用机制新药。 内分泌代谢领域再添重磅新药 替尔泊肽 2 型糖尿病和肥胖适应症均在国内获批 降糖减重领域明星靶点 GLP-1,在去年又迎来一个重磅产品,礼来的替尔泊肽首次在华获批上市。这是目前全球首个且唯一获批的 GIPR/GLP-1R 双靶点激动剂新药,在去年 5 月 和 7 月,其 2 型糖尿病和肥胖适应症相继在中国获批。 在减肥赛道爆火的当下,替尔泊肽的出现无疑让该赛道再起硝烟,同样作为 GLP-1 药物,势必要和早已声名鹊起的司美格鲁肽相提并论。在一项替尔泊肽和司美格鲁肽头对头的 Ⅲ 期研究中(SURMOUNT-5),替尔泊肽在第 72 周的平均减重效果(-20.2%)显著优于司美格鲁肽(-13.7%)。 国产 PCSK9 单抗添新将 降脂明星靶点 PCSK9 在 23 年迎来首个国产单抗获批后(信达的托莱西单抗),24年又有 2 款国产品种走向台前,分别是君实生物的昂戈瑞西单抗和康方生物的伊努西单抗。此外,恒瑞的瑞卡西单抗也在今年 1 月 10 日获批。 至此,国内已经有 7 款靶向 PCSK9 的产品获批上市,包括 6 款单抗,1 款 siRNA 药物。 国产自免药物大爆发 2024 年国产自免药物迎来密集收获期,有 3 款获批上市,分别是在同一天获批的 IL-17A 单抗(恒瑞医药夫那奇珠单抗、智翔金泰赛立奇单抗),以及康诺亚的 IL-4R 单抗司普奇拜单抗。 上述 3 款药物的获批,标志着国产自免药物的崛起,打破了既往外资药企在该领域的垄断地位。 另外,纵观当下 IL-17A 和 IL-4R 单抗市场,不难发现,IL-17A 单抗竞争更为激烈,全球已有 6 款药物上市,1 款处于申请上市,如何赢得更多市场份额成为制胜关键。 反之,IL-4R 药物领域,全球仅有两款药物上市(即赛诺菲/再生元的度普利尤单抗、康诺亚的司普奇拜单抗),当下竞争相对缓和。但值得注意的是,已有 8 款国产 IL-4R 单抗处于临床 Ⅲ 期阶段,放眼未来,该领域的竞争激烈程度同样不可小觑。 国产双抗迎来重大突破 去年 5 月,康方生物自主研发的 PD-1/VEGF 双特异性抗体依沃西单抗获 NMPA 批准上市,用于联合培美曲塞和卡铂,治疗经 EGFR-TKI 治疗后进展的 EGFR 基因突变阳性的局部晚期或转移性非鳞 NSCLC。 依沃西单抗不仅是全球第一个获批上市的「肿瘤免疫+抗血管生成」机制的双特异性抗体,也是中国第二个独立自主开发获批上市的双特异性抗体新药。 除已经获批的 EGFR 突变晚期非鳞 NSCLC 后线人群的适应症外,更让业界眼睛一亮的是,依沃西单抗在 PD-L1 阳性晚期 NSCLC 一线治疗的 Ⅲ 期临床中取得成功(HARMONi-2 研究),成为全球首个单药头对头击败「药王」 K 药的药物。凭借该结果,康方生物在 2024 年 7 月向 NMPA 递交了上市申请,Insight 数据库预测有望在 25 年 Q1 获批,届时依沃西单抗将改变该领域长期以 PD-1 单抗为主的治疗格局,具有里程碑意义。 跨国药企持续发力 从获批数量上看,24 年获批的新药仍以进口居多,其中多家跨国药企的全球首创新药在中国获批。 比如安斯泰来的全球首创靶向 Nectin-4 的 ADC 药物维恩妥尤单抗、强生全球首个靶向 BCMA 和 CD3 的双抗特立妥单抗、礼来全球首个非共价、可逆 BTK 抑制剂匹妥布替尼,以及百时美施贵宝全球首创 Myosin 抑制剂玛伐凯泰等重磅新药均在国内获批,为国内相关患者带来新的治疗选择。 前衍可供研究的部分化合物 中文名 英文名 CAS号 链接 奥希替尼 Osimertinib 1421373-65-0 订购 替尔泊肽 Tirzepatide 2023788-19-2 订购 恩替司他 Entinostat 209783-80-2 订购 依匹哌唑 Brexpiprazole 913611-97-9 订购 玛伐凯泰 Mavacamten 1642288-47-8 订购 西司他丁 Cilastatin 82009-34-5 订购 亚胺培南 Imipenem 64221-86-9 订购 佐利替尼 Zorifertinib 1626387-80-1 订购 瑞普替尼 Ripretinib 1442472-39-0 订购 米洛巴林苯磺酸盐 Mirogabalin besylate 1138245-21-2 订购
2025-01-21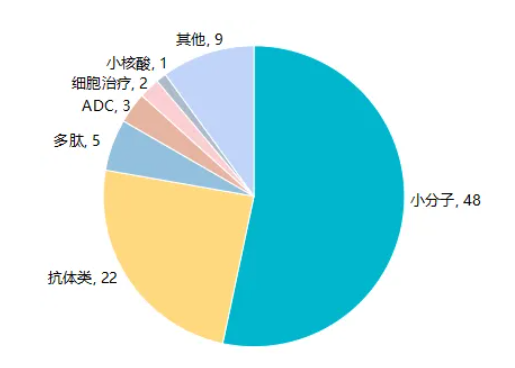
2025年全球畅销药前十预测,GLP-1产品独占4款
12月10日,《Nature Reviews Drug Discovery》期刊发表了2025年全球畅销药TOP10预测。前三名并不意外——Keytruda(帕博利珠单抗:俗称“K药”)、Ozempic(司美格鲁肽:减重领域爆火)和Mounjaro(替尔泊肽:同样减重领域爆火),与今年前三季度的销售表现吻合。值得注意的是,有4款GLP-1产品进入了预测名单,他们都属于代谢治疗领域。此外,畅销药榜单常客Dupixent(度普利尤单抗)和Skyrizi(利生奇珠单抗)两款自免药物也仍在榜单中,位居4、5位。 2025年全球畅销药TOP10(亿美元) 序号 通用名 商品名 公司 靶点 适应症 销售额预测 1 帕博利珠单抗 Keytruda 默沙东 PD-1 肿瘤 309.9 2 司美格鲁肽 Ozempic 诺和诺德 GLP-1R 代谢 222.5 3 替尔泊肽 Mounjaro 礼来 GLP-1R/GIPR 代谢 197.9 4 度普利尤单抗 Dupixent 赛诺菲 IL-4Rα 自免 169.4 5 利生奇珠单抗 Skyrizi 艾伯维 IL-23 自免 137.2 6 阿哌沙班 Eliquis BMS Factor Xa 心血管 135.2 7 比克恩丙诺 Biktarvy 吉利德 HBV polymerase/ HIV-1 integrase/ HIV-1reverse transcriptase 感染 134.3 8 司美格鲁肽 Wegovy 诺和诺德 GLP-1R 代谢 133.6 9 达雷妥尤单抗 Darzalex 强生 CD38 肿瘤 131.7 10 替尔泊肽 Zepbound 礼来 GLP-1R/GIPR 代谢 113.1 适应症覆盖广,“K药”有望再次成为“药王” 凭借着对肿瘤适应症的全面覆盖,Keytruda(帕博利珠单抗)在肿瘤领域一路狂奔,2023年的全球销售收入已达到250.11亿美元,首次登顶全球畅销药第一,成为新的“药王”,并且与第二名Humira(阿达木单抗)拉开了近百亿美元的差距。2024年上半年,“K药”再次登顶“药王”,其全球销售额为142.17亿美元。默沙东财报显示,2024年前三季度,帕博利珠单抗贡献了216.46亿美元,同比增长18%。 2023年全球畅销药TOP10(亿美元) 序号 药品 公司 适应症 2023年销售额 1 Keytruda (帕博利珠单抗) 默沙东 肿瘤 250.11 2 Humira (阿达木单抗) 艾伯维 自免 144.04 3 Ozempic (司美格鲁肽) 诺和诺德 代谢 139.17 4 Eliquis (阿哌沙班) BMS/辉瑞 心血管 122.06 5 Biktarvy (比克恩丙诺) 吉利德 感染 118.5 6 Dupixent (度普利尤单抗) 赛诺菲/再生元 自免 115.88 7 Comirnaty (新冠疫苗) 辉瑞/BioNtech 感染 112.2 8 Stelara (乌司奴单抗) 强生 自免 108.58 9 Jardiance (恩格列净) BI/礼来 代谢 107.5 10 Opdivo (纳武利尤单抗) BMS/小野 肿瘤 100.35 自2014年上市以来,K药已在全球范围内获批40个适应症,这些适应症涵盖了多种癌症类型。Keytruda的适应症探索仍未触及边界,自2023年至今又陆续拿下了宫颈癌等10项新的适应症。随着默沙东对其适应症及其治疗组合的持续探索,以及明年有望上市的帕博利珠单抗皮下注射剂型带来的助力,Keytruda的销售额仍然会继续爬升。根据Evaluate副总裁Paul Verdin的预测,其销售峰值可达到310亿美元。 Keytruda自2023年至今新增的适应症 获批时间 适应症 2023/1/27 作为手术切除且铂类化疗后的IB期(T2a≥4厘米)、II期或IIIA期非小细胞肺癌 (NSCLC)成年患者的辅助治疗 2023/3/28 不可切除或转移性MSI-H或dMMR实体瘤的成年和儿童患者 2023/10/16 联合含铂化疗作为可切除(肿瘤≥4厘米或淋巴结阳性)非小细胞肺癌(NSCLC)患者的新辅助治疗,单药作为手术后的辅助治疗 2023/11/1 联合吉西他滨和顺铂治疗局部晚期不可切除或转移性胆道癌(BTC)患者 2023/11/16 联合化疗作为局部晚期不可切除或转移性人表皮生长因子受体2(HER2)阴性胃癌或胃食管交界处癌成年患者的一线治疗 2023/12/15 联用维恩妥尤单抗治疗局部晚期或转移性尿路上皮癌成人患者 2024/1/12 联合化疗治疗FIGO(国际妇产科联合会)III-IVA期宫颈癌患者 2024/1/25 既往接受过除含PD-1/PD-L1方案以外的全身治疗的乙型肝炎继发性HCC患者 2024/6/17 先联合卡铂和紫杉醇,随后单药治疗原发性晚期或复发性子宫内膜癌成年患者 2024/9/18 联合培美曲塞和铂类化疗作为不可切除的晚期或转移性恶性胸膜间皮瘤(MPM)成年患者的一线治疗 减重领域迎来爆发,多款GLP-1产品上榜 近两年,GLP-1赛道的盛况有目共睹,已经有4款产品进入2023年全球畅销药TOP100名单,其中3款产品为不同商品名的司美格鲁肽。若是按通用名计算,司美格鲁肽与帕博利珠单抗的2023年总销售额差距并不大(212 vs 250亿美元),并且双方差距在今年前三季度已经大幅缩小(206 vs 216亿美元)。另一款明星GLP-1产品Mounjaro/Zepbound(替尔泊肽)同样表现非凡,获批上市仅两年,销售额便迈过50亿美元门槛,今年还将轻松闯过百亿美元大关。 GLP-1作为非胰岛素类降糖药开发的三大热门靶点之一,近年来在减重药物领域也展现出了巨大的潜力和广阔的行业前景。此次有4款产品进入预测榜单,其中两款产品为诺和诺德不同商品名的司美格鲁肽;两款为礼来不同商品名的替尔泊肽。根据预测名单,Ozempic、Wegovy、Mounjaro和Zepbound这4款GLP-1产品在2025年将创造合计667亿美元的收入,占TOP10产品销售总额的40%。 在司美格鲁肽和替尔泊肽的带动之下,GLP-1靶点的研发热度也迅速到达了顶点。GLP-1之热至少有一半原因来自于庞大的减肥需求,正规产品稀缺的减肥市场孕育着极大的红利,任谁都想分一杯羹。未来,随着小分子GLP-1和多靶点产品逐渐进入市场,GLP-1市场的厮杀会更加激烈。 自免产品也是全球畅销药榜单的常客 Paul Verdin认为明年的榜单前5名将包含Dupixent(度普利尤单抗)和Skyrizi(利生奇珠单抗)两款自免药物。 在攻克慢阻肺病(COPD)之后,Dupixent(度普利尤单抗)的市场潜力来到了新的高度,今年这款产品将达到赛诺菲自己设立的130亿欧元销售峰值目标。Paul Verdin也认可Dupixent(度普利尤单抗)的实力,预测其2025年销售额将达到169.4亿美元。 Humira虽迟暮,但艾伯维已带着Skyrizi杀回免疫霸主卫冕之战。目前,利生奇珠单抗处于狂揽自免适应症阶段,斑块状银屑病、银屑病关节炎、克罗恩病和溃疡性结肠炎已被其拿下。Skyrizi(利生奇珠单抗)的2023年销售额为77.63亿美元,位列全球畅销药榜单第15位。从今年各家产品前三季度的情况来看,在Comirnaty(新冠疫苗)和Opdivo(纳武利尤单抗)被挤出前十之后,Skyrizi可能卡位第10。 总体而言,2025年预测名单中仅2款小分子药物上榜,余下8款为多肽药物和生物大分子药物。在往年的全球畅销药物TOP10榜单中,小分子药物和生物大分子药物的席位基本持平。随着几款小分子药物在近几年陆续失去专利保护,生物大分子药物开始主导TOP10榜单。在GLP-1药物的销售额出现爆发式增长之后,明年的榜单也将发生新的变化。 前衍可以提供的用于实验的原料化合物 中文名 英文名 CAS号 链接 帕博利珠单抗 Pembrolizumab 1374853-91-4 订购 司美格鲁肽 Semaglutide 910463-68-2 订购 替尔泊肽 Tirzepatide 2023788-19-2 订购 阿哌沙班 Apixaban 503612-47-3 订购 阿达木单抗 Adalimumab 331731-18-1 订购 达雷妥尤单抗 Daratumumab 945721-28-8 订购 乌司奴单抗 Ustekinumab 815610-63-0 订购 恩格列净 Empagliflozin 864070-44-0 订购 纳武利尤单抗 Nivolumab 946414-94-4 订购
2025-01-15
Nature Medicine:将影响2025年医学的11项临床试验
2024年的大赢家一定是减肥药索马鲁肽(诺和诺德的 Wegovy)和替尔泊肽(礼来的 Zepbound)背后的制药公司。随着其他生物技术和制药公司纷纷进入减肥市场,肥胖症必将成为未来很长一段时间的热点。那么在肥胖之外,还有什么领域值得关注、什么新药值得期待呢? 2024年12月12日,自由撰稿人Paul Webster和Natalie Healey在Nature Medicine 杂志上发表年度回顾文章Eleven clinical trials that will shape medicine in 2025。通过采访11位前沿科学家,列出了2025年的11项重磅临床试验,从朊病毒病和镰状细胞病的基因疗法到癌症和精神健康的数字工具。 朊病毒病的基因治疗 提名人:Sonia Vallabh,来自美国麻省理工学院/哈佛大学博德研究所 这位提名人是位为了自救,半路出家弃律从医(并且只用了7年就从0到博德研究所的独立实验室)的女科学家。 朊病毒病是一种由朊病毒引起的传染性脑病,主要影响人类和动物的中枢神经系统。朊病毒是一种特殊的蛋白质,能够自我复制并传播疾病。这种疾病的特征是进行性脑功能紊乱,无缓解康复,终至死亡,且死亡率为100%。 Vallabh团队开发的用于治疗朊病毒病的全新表观遗传编辑技术此前发表于《科学》杂志。此次她提名的正是一种针对朊病毒病的鞘内给药反义寡核苷酸ION-717的1/2期临床试验。目前,该研究已经在全球16个中心开展,有望在2025年底获得第一批可供分析的数据。 多元成人队列中的精准营养学探索 提名人:Leanne Redman,来自美国彭宁顿生物医学研究中心 一种健康食物,到底能够给人提供多少“健康”?这个问题的答案其实个体差异很大。就算有严格设计的临床试验,但受限的参与者特征很难完全概述食物的健康收益。 Redman提名的是一项针对大规模、多样化受试者群体进行的营养学研究,参与者超过8000名成年人,几乎不设排除标准,在他们日常的药物和健康状况的背景下,能最大程度地预测不同饮食模式对不同人群的健康影响。这项研究将于2026年夏季完成,2025年初或有前期数据披露。 CBD预防精神病 提名人:Philip McGuire,来自牛津大学 McGuire提名的是一项名为早期精神疾病分层和治疗(STEP)的研究,该研究使用已经获批用于治疗严重癫痫的大麻二酚(CBD),评估其在精神疾病中的疗效。 该研究包括三项临床试验,涉及11个国家/地区的30个中心的1000名精神疾病患者,通过神经影像学评估疗效,并尝试寻找可用的生物标志物。 该研究亮点在于囊括了处于不同疾病阶段的患者,包括高风险但未发病人群、新发病人群、治疗耐药人群。该研究预计在2025年获得CBD初步的疗效、安全性和耐受性的数据,其结果将决定CBD是否具有预防精神疾病的潜力。 镰状细胞病的碱基编辑 提名人:刘如谦(David Liu),来自哈佛大学Merkin医疗保健转化技术研究所/博德研究所 刘如谦团队开发的碱基编辑(base editing)在基因治疗上已经取得了一定的成果,目前正在开展的是首个针对造血干细胞(HSC)的碱基编辑临床试验,这项试验的结果有望影响多种血液疾病的治疗。 这项名为BEACON的研究是一项开放标签、单臂、多中心、1/2期研究,评估体外碱基编辑的HSC在严重镰状细胞病患者中的安全性和有效性。该研究预计在2025年初获得初步结果。 通过给屋顶降温防止热病 提名人:Aditi Bunker,一位流行病学家 近年来极端气候频发,尤其是夏季极端高温,造成了不少疾病和死亡。Bunker提名的是一项在西非地区进行的使用高反射率屋顶涂料降低室内温度的研究,该研究随访了600个家庭1200个个体2年间热暴露与多种身体指标的关系。这将有助于改善受严酷高温影响地区的人们的生活。 前列腺癌的放射性核素疗法 提名人:Oliver Sartor,来自梅奥诊所肿瘤科 Lu177作为前列腺癌药物在近年才取得进展。FDA已经批准其生物制剂用于治疗前列腺特异性膜抗原(PSMA)阳性的转移性去势抵抗性前列腺癌(mCRPC)成人患者。 有几项研究正在探索是否可以在更早阶段开启Lu177治疗,如已经报告结果的PSMAfore。2025年FDA将根据这项试验结果决定Lu177是否获得新的适应症。 使用AI进行宫颈癌筛查 提名人:Farida Selmouni,来自法国里昂国际癌症研究机构(IARC) 法国的全国宫颈癌筛查计划包括对未在诊所接受筛查的30-65岁女性进行HPV自我采样,但实际参与率不足20%。 Selmouni提名的项目是基于网络的多语言决策辅助工具(AI聊天机器人),针对受教育程度较低和来自弱势地区的女性进行了优化,可通过智能手机访问,为使用者提供相关的知识服务,以便提高筛查计划的参与率。 目前该项目正在进行随机对照试验,预计于2025年结束。 心理健康移动工具包(mSELY) 提名人:Keng-Yen Huang,来自纽约大学医学院 这项研究旨在解决低收入和中等收入国家青少年所面临的发育、性、生殖健康等心理健康障碍问题,通过帮助青少年建立同龄人联系的同时自我评估和管理,该项目也面向青少年父母提供心理健康教育支持。 个性化乳腺癌筛查 提名人:Suzette Delaloge,来自法国古斯塔夫·鲁西研究所(IGR) 世界上大多数国家的乳腺癌筛查策略基于年龄,从女性50岁开始执行筛查,但仅将死亡率降低了20%,且出现了较高比例的过度诊断和不必要治疗。 根据女性自身风险因素的差异,如遗传、生活方式、激素暴露,实际上可以灵活修改筛查策略。Delaloge提名的研究是目前同类研究中规模最大的,在6个国家的53000余名女性中进行,对比标准乳腺癌筛查和个性化筛查策略的优劣。 家庭菜园改善气候相关营养不良 提名人:Ina Danquah,来自德国波恩大学 Danquah提名的这项研究,是尝试通过家庭菜园解决气候变化导致的主食作物营养物质水平降低问题,以参与研究的家庭中儿童的营养状况为主要评估标准。 通过手机游戏改善自闭症儿童症状 提名人:Dennis P.Wall,来自斯坦福大学医学院 自闭症儿童存在社交方面的障碍,早期干预能一定程度上改善这些问题。该提名研究设计了一款手机游戏,可以有目的地针对社交期间眼神接触、动机、注意力等要点进行训练,并有效减少焦虑,有希望减轻儿童父母的压力、尽量减少不利于儿童融入社会的自闭症状。该游戏收集到的数据也可以用于训练人工智能工具,帮助自闭症群体。 前衍可以提供用于临床实验的原料 中文名 英文名 CAS号 链接 司美格鲁肽 Semaglutide 910463-68-2 订购 替尔泊肽 Tirzepatide 2023788-19-2 订购
2025-01-09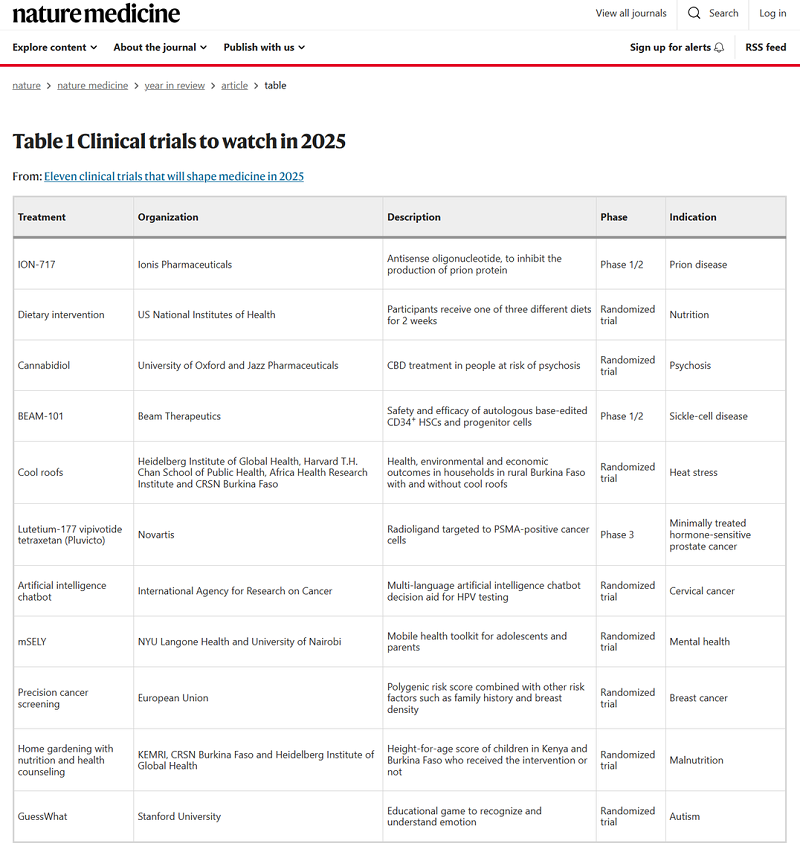
六起危化品相关违法处罚案例
案例1:浙江某包装材料有限公司未落实安全管理措施的行政处罚案 2024年8月8日,临海市应急管理局接市民来电举报,对浙江某包装材料有限公司进行安全生产执法检查,发现该公司2#丙类仓库二楼存放有聚氨酯胶黏剂(编号为FD-8960)约13吨,聚氨酯胶黏剂(编号为FD-3160) 约3吨,以上物料属于危险化学品(但不属于剧毒化学品),未存放在危险化学品专用仓库内。执法人员当场依法下达《现场处理措施决定书》(临应急现决[2024]20-35号)。8月12日,临海市应急管理局对浙江伟涛包装材料有限公司涉嫌的安全生产违法行为予以立案。8月14日,对该公司总经理及有关人员进行了调查询问,调阅有关资料,提取有关凭证。8月22日结束案件调查。 经核查,浙江某包装材料有限公司存在未将危险化学品储存在专用仓库内的违法行为,其行为违反了《危险化学品安全管理条例》第二十四条第一款“危险化学品应当储存在专用仓库、专用场地或者专用储存室(以下统称专用仓库)内,并由专人负责管理”之规定,依据《危险化学品安全管理条例》第八十条第一款第(四)项之规定,2024年9月11日,临海市应急管理局决定给予浙江伟涛包装材料有限公司罚款人民币6万元的行政处罚。 案例2:浙江某安全科技有限公司安全评价报告存在法规标准引用错误的重大疏漏行政处罚案 2024年6月26日,台州市仙居县应急管理局依法对浙江某安全科技有限公司在仙居的安全评价报告进行安全检查时,发现该公司编制的浙江某药业转型升级项目设立安全评价报告存在多处法规标准名字书写错误,引用的《石油化工控制室抗爆设计规范》、防盗安全门应符合的GB17565-2007等多处法规标准过期等问题。当日,仙居县应急管理局对该公司的违法行为立案调查。 经调查,浙江某安全科技有限公司为浙江某药业转型升级项目作出的安全评价报告安全评价报告存在法规标准引用错误的行为违反了《安全评价检测检验机构管理办法》第二十二条第(五)项“安全评价检测检验机构及其从业人员不得出具虚假或者重大疏漏的安全评价、检测检验报告”之规定,依据《安全评价检测检验机构管理办法》第三十条第(十)项之规定,2024年7月8日,仙居县应急管理局决定对浙江某安全科技有限公司作出罚款人民币0.5万元的行政处罚 案例3:淄博一男子非法制售危险化学品获刑 近日,博山区人民法院对某化工厂、张某某(化名)非法经营一案进行公开审理并当庭宣判,被告单位某化工厂犯非法经营罪,判处罚金人民币四十五万元,被告人张某某犯非法经营罪,触犯了《中华人民共和国刑法》第二百二十五条第(一)项“未经许可经营法律、行政法规规定的专营、专卖物品或者其他限制买卖的物品的”,判处有期徒刑二年六个月,缓刑三年六个月,并处罚金人民币十五万元。 被告人违反国家规定,在其经营的化工厂内,未经许可私自仿制生产危险化学品试剂并对外进行销售。经查明,该厂向外销售危险化学品试剂共18722瓶,销售金额共计1415414元。被告单位违反国家规定,未经许可经营法律、行政法规限制买卖的物品,扰乱市场秩序,情节严重,其行为已构成非法经营罪。被告人张某某作为被告单位直接负责的主管人员,依法应当承担相应的刑事责任。被告单位某化工厂、被告人张某某有视为自首情节,自愿认罪认罚,依法对其从轻处罚并适用缓刑。 案例4:湖北一男子非法经营“笑气”获刑 2023年1月至8月,彭某在未办理危险化学品经营许可证的情况下,多次以每罐80元的价格从他人手中购买罐装危险化学品一氧化二氮(俗称“笑气”),后以每罐100元到150元的价格进行贩卖。2023年8月,彭某在向周某销售2罐“笑气”时被抓获。 仙桃法院经审理认为,被告人彭某违反国家规定,未经许可经营国家法律、行政法规规定的限制买卖的危险化学品,扰乱市场秩序,情节严重,其行为构成非法经营罪。仙桃法院依法判决彭某犯非法经营罪,,触犯了《中华人民共和国刑法》第二百二十五条第(一)项“未经许可经营法律、行政法规规定的专营、专卖物品或者其他限制买卖的物品的”,判处有期徒刑一年,并处罚金人民币二万元。 “笑气”的化学名称为一氧化二氮,有轻微麻醉作用,因吸入后会感到轻快,能致人发笑而得名。“笑气”对人体的危害与毒品相似,具有成瘾性,滥用对身体危害极大。尽管“笑气”目前尚未被作为毒品进行列管,但作为危险化学品,非法贩卖、吸食“笑气”同样受到法律规制。 吸食“笑气”可能会因“非法使用危害物质”或“非法持有危害物质”受到行政处罚,违规生产、销售“笑气”可能会构成非法经营罪。广大群众应提高法律意识和辨别能力,不好奇、不尝试、不滥用,如发现不法分子贩卖“笑气”,需及时举报。 案例5:姚某华非法经营危险化学品(柴油)案 【基本案情】 接群众举报,2024年4月19日,涟源市应急管理局对荷塘镇观桥村一工地进行安全检查,发现工地上一辆设置储油罐和加油设施的厢式货车正向一挖掘机加注危险化学品(柴油)。经查,设置储油罐和加油设施的厢式货车系姚某华所有,姚某华未取得《危险化学品经营许可证》,2024年2月,将购买的厢式货车改装成具有储存、加油功能的流动加油装备,为自己挖掘机加油的同时,2024年3月起,先后11次向从事挖掘作业的同行销售危险化学品(柴油),至查获之日,非法所得1.0556万元。 【处理情况】 姚某华非法经营危险化学品(柴油)的行为,违反《危险化学品安全管理条例》第三十三条第一款,依据《危险化学品安全管理条例》第七十七条第三款之规定,涟源市应急管理局作出责令其立即停止非法经营活动,没收违法所得1.0556万元,处10.1万元罚款的行政处罚。 【典型意义】 柴油属于易燃易爆危险物品,发生火灾或爆炸概率高,危险性大。国家对危险化学品的经营实行许可证制度,未经许可,任何单位或者个人不得经营危险化学品。本案当事人为逃避监管,打着自用幌子,非法进行危险化学品销售,严重危害公共安全和经营秩序。本案的严厉查处,有利于维护安全生产行政执法的权威,起到“查处一个、震慑一批、教育一片”的效果。 案例6:王某勇非法生产危险化学品(固体酒精蜡)案 【基本案情】 接群众举报,2024年1月5日,花垣县应急管理局对花垣镇花桥村一闲置厂房进行安全检查,发现有3人正在进行生产作业,现场存放用作火锅燃料的绿色包装固体酒精蜡块5450件、红色包装固体酒精蜡块4893件、片状氢氧化钠37包、硬脂酸68包、酒精半罐、成品固体酒精591瓶。抽样经检测机构检测,固体酒精蜡块主要成分为甲醇和乙醇,属危险化学品的有机易燃固体。经查,王某勇未取得《危险化学品安全生产许可证》,自2022年6月以来,以每年2.5万元租用花垣县花垣镇花桥村一闲置厂房,购买甲醇、乙醇、硬脂酸、氢氧化钠等作为原料,雇佣3名工人,生产固体酒精蜡块。至查获之日,已非法获利5.0715万元。 【处理情况】 王某勇非法生产危险化学品的行为,违反了《安全生产许可证条例》第二条的规定。依据《安全生产许可证条例》第十九条之规定,2024年5月31日,花垣县应急管理局对其作出责令停止生产,没收违法所得5.0715万元,处15万元罚款的行政处罚。 花垣县应急管理局依据有关规定,给予举报人1万元奖励。 【典型意义】 甲醇属于易燃易爆化学品,是国家重点监管和特别管控的物品,其蒸气与空气可形成爆炸性混合物,遇明火、高热极易引起燃烧爆炸,危险性较高。本案涉案人员心存侥幸,未经许可,租用闲置厂房非法生产固体酒精蜡块,生产现场设备简陋,无安全设施和消防设施,极易发生事故。执法人员通过调查询问、抽样送检等锁定证据,依法予以严厉处罚,有力打击了危险化学品非法生产行为,消除了安全隐患。
2025-01-03
稀土材料:医疗领域大放光彩
稀土材料作为一类金属材料,拥有独特的物理性质和化学性质。近年来,稀土材料在医学领域的应用日益广泛。 从疾病早期诊断到精准治疗,稀土材料正在助力医疗水平的进一步提升。 多种诊疗技术“强助攻” 稀土是元素周期表中镧系(镧、铈、镨、钕、钷、钐、铕、钆、铽、镝、钬、铒、铥、镱、镥)15个元素和21号元素钪、39号元素钇共17个元素的总称。一直以来,科学家不断探索稀土元素的生物学效应。早在20世纪50年代,诺贝尔化学奖得主、美国化学家格伦·西奥多·西博格及其团队就通过实验验证了某些稀土化合物的安全性,并发现它们对生物具有独特效用。 稀土元素拥有特殊的电子层结构,能够有效吸收和发射特定波长的光,从而能与其他类型材料结合,形成性能各异、品种繁多的新型光功能材料。这一特性为其在医学影像学中的应用提供了理论支撑。同时,部分稀土离子具有良好的稳定性和与生物分子结合的能力,为相关药物设计开辟了新途径。 随着稀土分离技术进步,自20世纪60年代以来,稀土元素开始应用于医学领域。在医学治疗方面,以钆为基础的造影剂广泛应用,极大提高了图像对比度和诊断准确性;含有铕的荧光探针,则被用于细胞追踪和生物标志物检测;钐对于缓解骨转移癌患者疼痛,也能起到显著效果。 基于稀土近红外二区发光的前哨淋巴结转移精准成像技术。该技术不仅能让癌细胞更早被发现,还能在手术中精准定位癌细胞,为医生“导航”。 此外,稀土在提高生物医用材料性能方面有得天独厚的优势。例如铈掺杂的磷酸钙可被用于骨骼修复,镧系元素掺杂的二氧化钛表面拥有优异的抗菌性能。不仅如此,陆续被发现的稀土化合物还具有一系列特殊药效作用,可用于治疗烧伤、炎症、皮肤病、血栓病等。 利用稀土材料有望研发出像导弹一样的靶向药物。有科学家正在研究采用稀土中的镥、钇等放射性同位素精准定位肿瘤细胞,再利用其放射性特质发射出高能粒子,定向杀死肿瘤细胞。 取得诸多研究成果 提高药物的生物利用率、靶向定位准确度,以及降低毒副作用,一直是药物载体领域的研究重点。基于稀土元素开发的多功能复合材料作为药物载体,能精确控制药物在体内的释放,实现药物缓释,在提升疾病治疗效果方面展现出较大潜力。 不久前,中国科学院福建物质结构研究所研究员卢珊、陈学元等研发出新型稀土纳米光学诊疗材料,在肺部感染小鼠模型中实现了非侵入性光学诊疗。研究人员介绍,这种新型材料采用独特的富集—包覆策略,将数百个超小纳米颗粒封装在介孔二氧化硅内核中,形成尺寸约220纳米的均匀、单分散性好、介孔负载量大的纳米杂化材料。其作为药物载体更易递送到肺部,提高了生物利用度。 此外,以活体荧光成像法替代癌症病理诊断使用的活检样本体外检测技术,成为一种新型无创肿瘤病理诊断方法,受到临床医生青睐。其中,成像的关键一环离不开稀土。 稀土材料之所以能在成像方面表现突出,主要得益于其优异的光学特性、高稳定性、多功能性以及深组织穿透能力等。厦门大学附属第一医院核医学科副主任医师、教授陈皓鋆介绍:“稀土元素特别是某些稀土同位素,如镥-177,具有独特的物理和化学性质,非常适合用于癌症靶向治疗和成像。” 以上述基于稀土近红外二区发光的前哨淋巴结转移精准成像技术为例,该研究团队负责人、中国科学院海西研究院厦门稀土材料研究中心研究员张云介绍,他们研发的这种神奇的纳米探针并非一种针状物体,而是一种化学合成物质。研究团队利用近红外Ⅱb区发光的稀土下转换纳米颗粒作为发光元件,以肿瘤特异性靶点CXCR4的拮抗肽作为靶向元件,成功合成出新型近红外Ⅱb区纳米探针。在临床应用时,只需注射这种会发光的纳米探针,即可自动定位肿瘤;通过光学成像仪器,就能隔着皮肤和肌肉组织直观清晰观察到肿瘤的边缘、细胞的游走。 为了提升肿瘤靶向能力,陈皓鋆团队尝试利用稀土纳米材料多光谱发射的特性,设计合成多色、多指标肿瘤免疫微环境精准识别探针,开展纳米刀治疗下肿瘤免疫微环境的多指标精准动态识别研究,实现对肿瘤免疫微环境的多层次多指标精准动态检测。“目前,我们已经搭建了纳米刀治疗平台,正在推进后续基于稀土纳米探针的纳米刀消融治疗诱发机体免疫效应动态识别研究。”陈皓鋆说。 加快推动应用落地 稀土材料在医学领域的应用并非没有挑战。如何确保这些材料在人体内安全使用,避免潜在生物毒性和不良反应,是科学家们必须面对的问题。此外,稀土材料的提取和制造过程相对复杂,成本较高,这也限制了其在一些资源有限地区的普及。 推动更多安全可靠的稀土应用在医学领域落地,无疑需要大量扎实的基础试验数据。为此,一些科研团队及医疗机构纷纷布局,致力于探索稀土在医学领域的新用途,期望通过开发更多稀土新型材料,提升疾病诊断准确性与治疗有效性。 2022年,长期从事无机化学研究的中国科学院院士洪茂椿与厦门大学附属第一医院合作成立中国稀土学会稀土医学临床研究中心,携手开展医工交叉创新合作,启动稀土元素在医学诊疗技术上的系列研究。 洪茂椿表示,稀土的独特功能在生物医学领域有着广阔的应用前景,“我们希望研究出更多稀土离子或稀土化合物在生物体内的作用机理,用以开发医用材料和药物,实现疾病的精准诊疗。” 前衍可以提供的稀土材料 中文名 英文名 CAS号 链接 镧 Lanthanum 7439-91-0 订购 铈 CERIUM 7440-45-1 订购 镨 Praseodymium 7440-10-0 订购 钕 NEODYMIUM 7440-00-8 订购 钷 Promethium 7440-12-2 订购 钐 SAMARIUM 7440-19-9 订购 铕 EUROPIUM 7440-53-1 订购 钆 GADOLINIUM 7440-54-2 订购 铽 Terbium 7440-27-9 订购 镝 DYSPROSIUM 7429-91-6 订购 钬 Holmium 7440-60-0 订购 铒 ERBIUM 7440-52-0 订购 铥 THULIUM 7440-30-4 订购 镱 Ytterbium 7440-64-4 订购 镥 LUTETIUM 7439-94-3 订购 钪 Scandium 7440-20-2 订购 钇 Yttrium 7440-65-5 订购
2024-12-27
Nature2024十大科学人物!我国2位科学家上榜!
2024年12月9日,《自然》公布了2024年度十大人物榜单(Nature's 10)——这一榜单旨在选出十位在这一年重大科学事件中占有一席之地的人物。 年度十大科学人物是《自然》从当年一些宏大科学故事的参与者中评选出的10个人。《自然》特写部编辑布兰登·马赫表示:“今年的十大科学人物对科学界和全世界产生了不容忽视的影响。” 现代科学研究,往往是由团队,甚至是多个团队协作完成,但科研世界也充满了个人发挥影响的故事。《自然》年度十大人物就是这样,他们代表了影响2024年一些最重要科学事件的个人。从探索月球背面的深处到彻底改变自身免疫性疾病的治疗方法,这些开拓者突破了界限。 值得注意的是,凭借“一个”细胞,一捧土壤,今年有两位中国科学家入选。其一,为中国人民解放军海军军医大学内科医生徐沪济,他利用供体来源的基因编辑T细胞成功治疗了毁灭性的自身免疫性疾病。该疗法基于T细胞在癌症治疗中取得的成功,为前沿的CAR-T治疗的批量生产带来了希望。其二,则是中国科学院国家天文台研究员、嫦娥六号任务工程副总设计师李春来,他是首位亲手拿到嫦娥六号今年返回地球的月球土壤样本的科学家。 【徐沪济——大胆医生:首创 CAR-T 疗法治疗自身免疫性疾病】 《自然》评价徐沪济为Daring doctor,意为大胆医生,以表彰他及他的团队首创性的使用异体来源的CAR-T疗法治疗的自身免疫疾病患者。 2024年7月16日,徐沪济携手邦耀生物、华东师范大学及浙江大学医学院第二附属医院的研究同仁,在《Cell》杂志上发表研究成果。 徐沪济透露,首次治疗完成后,团队成员都心绪难宁,夜不能寐,因为所涉病例均为自身免疫疾病重症患者。 历经两周治疗,第一位患者(罹患免疫介导性坏死性肌病)肌力恢复,能自主抬臂;另两名男性患者(患弥漫性皮肤系统性硬化症)症状亦见减轻。历经六月有余,三位患者病情均获长期缓解,团队才稍感宽慰。 CAR-T细胞疗法(即嵌合抗原受体T细胞疗法)已是老生常谈,作为癌症免疫治疗的重大突破,自2017年起,已有六款获中国批准上市,其针对白血病、淋巴瘤等血液癌症疗效显著。 近年来,CAR-T疗法在系统性红斑狼疮、多发性硬化症等自身免疫疾病领域亦展现出了广阔前景。 然而,现有CAR-T疗法均基于患者自体免疫细胞进行基因改造,导致成本高昂且流程繁复,德国学者于2021年首例成功运用CAR-T疗法治疗系统性红斑狼疮,后续虽治疗多例自身免疫疾病患者,但均基于自体细胞。 鉴于此,中国研究团队开始探索利用健康捐赠者免疫细胞制备现货通用型CAR-T产品。理论上,单一捐赠者的细胞可治疗上百人,有望大幅提升生产规模、降低成本、缩短周期,进而增强CAR-T疗法的可及性。 邦耀生物团队从一名21岁女性志愿者的血液中提取T细胞,通过基因工程技术导入能识别并攻击CD19蛋白的基因,同时利用CRISPR-Cas9技术去除可能引起排斥反应的五个基因,再通过磁性珠子对目标物进行选择性分离以确保细胞纯净,最终成功研发出了新一代异体通用型CAR-T疗法。 这种疗法产量高,每批次产量足以满足100+患者需求(每次输注剂量为1×10^6/kg CAR阳性细胞),实现了安全有效的免疫细胞治疗。 在治疗后六个月内,三名患者(一名难治性免疫介导性坏死性肌病患者及两名弥漫性皮肤系统性硬化症患者)症状均深度缓解,临床反应指数显著改善,炎症及器官纤维化逆转,且未出现细胞因子释放综合征或其他严重不良反应。 这些临床数据表明,现货通用型CAR-T疗法在治疗难治性自身免疫疾病方面具有卓越的安全性和有效性。 其临床优势也展露无遗:患者可及性高、安全性强、疗效显著,有效解决了自体CAR-T疗法的高度个性化、制备周期长、失败风险高及成本昂贵等问题。 尤为值得一提的是,这三名中国患者是全球首批接受供体细胞(非自体细胞)来源CAR-T疗法治疗的自身免疫疾病患者,亦是《Cell》首次刊载CAR-T疗法治疗自身免疫疾病的研究成果,算得上是前无古人、后无来者了。 徐沪济还透露,另有24名自身免疫疾病患者接受了该疗法治疗,整体疗效积极。 华东师范大学生命科学学院/邦耀生物的杜冰教授作为该通用型疗法的共同开发者,对徐沪济的勇敢尝试表示高度赞扬。他透露,在寻求合作医生时,徐沪济是唯一敢于冒险尝试此新疗法的医生。 【李春来——月岩守护者】 2024年6月25日,中国嫦娥六号使命圆满,其返回舱满载着月球背面首批珍贵样本顺利归来。 中国国家天文台研究员、嫦娥六号项目副总设计师李春来,屏息凝视着返回舱缓缓降落,心中暗自祈愿:“样品,我终于得到你了。” 这一成就,不仅是对他多年心血的至高肯定,也标志着中国月球探索历程的新篇章正式开启。 月球背面隐藏在地球之外,至今仍是一个谜。嫦娥六号着陆器从古代南极艾特肯盆地收集了近两公斤的月球岩石,这是月球上最古老、最大的撞击坑之一。“这些样本就像数十亿年历史的记录者,”李解释道。这些新的见解可以揭示为什么月球的远端与近端不同,并为整个太阳系的行星演化提供线索。 此次任务的副总设计师、地质学家李春来负责监督选择着陆地点、分析样本以及协调由70多名科学家组成的团队的艰巨任务。“这些样本独具特色,”李春来细致描绘道,“它们质地轻盈、结构蓬松,色泽较月球正面样本更为浅淡。” 9月,李春来与其团队在《National Science Review》科学期刊上,发表了关于嫦娥六号返回样本的首篇研究论文,深入剖析了样本的物理、矿物及地球化学特性。 11月,他们的进一步研究成果在《Nature》与《Science》两大顶级科学期刊上同步发表。文中指出,这些样本中蕴含了月球背面火山活动持续至28亿年前的确凿证据。这一重大发现,瞬间吸引了全球科学界的瞩目。 而对于李春来而言,“今年月球任务所带来的科学发现,将仅仅只是个开端”。 不止两位中国科学家,另外八位上榜者也同样与他们的团队推动了惊人的发现: Rémi Lam:依托机器学习开发天气预报 AI 模型,实现更高效、更精准的全球天气预报。 Ekkehard Peik:实现核钟“首次滴答”,为下一代超高精度时间测量技术奠定基础。 Wendy Freedman:完善“标准烛光”校准方法,为解决宇宙膨胀率争议提供新线索。 Kaitlin Kharas:领导“支持我们的科学”运动,使加拿大的研究生和博士后获得了 20 年来最大的薪酬涨幅。 Anna Abalkina:揭露学术欺诈和劫持期刊,为全球学术生态的透明和诚信做出贡献。 Muhammad Yunus:带领孟加拉国过渡政府,推动国家治理改革与社会公平。 Placide Mbala:通过追踪和分析猴痘病毒,为新发传染病的应对提供科学依据。 Cordelia Bähr:通过气候诉讼赢得欧洲人权法院支持,将气候变化与人权保护紧密相连。
2024-12-20